呼吸道上皮细胞和肺的抗感染研究
Respiratory epithelial cells orchestrate pulmonary innate immunity
REVELATION:
雾化疗法 (vitamin C,NAC, surfactant, negative ions...)
Expression of hydrolase in lung tissues
1. Influenza Virus H5N1 Infection Can Induce ROS Production for Viral Replication and Host Cell Death
2. Influenza virus damages the alveolar barrier by disrupting epithelial cell tight junctions
3. Influenza-Induced Production of Interferon-Alpha is Defective in Geriatric Individuals, due to lower frequency of pDCs.
4. All interferons (alpha, beta and gamma) inhibit viral replication by interfering with the transcription of viral nucleic acid.
5,Omega 3 Fatty Acids May Reduce Bacterial Lung Infections Associated with COPD
6. High-dose vitamin C has therapeutic efficacy on acute pancreatitis. The potential mechanisms include promotion of anti-oxidizing ability of AP patients, blocking of lipid peroxidation in the plasma and improvement of cellular immune function.
7. Low density lipoprotein (LDL) inhibits endothelium-dependent relaxation. Vitamin C reduced the inhibitory effect of LDL.
8. mechanical ventilation, particularly where significant overstretch occurs, may drive the pathogenesis of fibrosis in patients with ARDS.
9.AMPK activator, metformin, restored their phagocytic capacity to uptake both apoptotic neutrophils and NETs.
10. sentinel epithelial cells and innate immune cells might be essential components of pathogenesis,
11.A major cause of respiratory failure during influenza A virus (IAV) infection is damage to
the epithelial–endothelial barrier of the pulmonary alveolus.12.AECs produces inflammatory cytokines,like interferons,defensins,and chemokines in reponse to viral infections
13. MSCs from bone marrow can transfer mitochondria to alveoli macrophage
14. Respiratory epithelial cells orchestrate pulmonary innate immunity
15. The AEC-induced migration of blood monocytes could be reduced by 30% to 90% by neutralizing MCP-1. AEC secrete high levels of MCP-1 and the murine IL-8 homologs KC and MIP-2 upon stimulation with LPS (20).
16. AEC II are cuboidal cells that constitute around 15% of total lung cells and cover about 7% of the total alveolar surface. AEC II are responsible for epithelium reparation upon injury and ion transport. AEC II contribute also to lung defense by secreting antimicrobial products such as complement, lysozyme, and surfactant proteins (SP). AEC II secrete a broad variety of factors, such as cytokines and chemokines, involved in activation and differentiation of immune cells and have been described to be able to present antigen to specific T cells (6–11).
17. activation of apoptosis and necroptosis pathways in monocytes differentially contributed to the immune response of monocytes upon H7N9 infection
18.IFNs-I were found to systemically activate natural killer (NK) cell activity and activity of other cells of the immune system, such as antigen-presenting dendritic cells (DC) and CD4 and CD8 T cells.
19.Respiratory Epithelial Cells as Master Communicators during Viral Infections
20.AT2 cells are the major cell type that produce GM-CSF during IAV infection and epithelial cell-produced GM-CSF contributes to disease attenuation and survival
21.IAV has also been shown to upregulate PD-L1 expression in human airway epithelial cultures. IAV-induced expression of PD-L1 is dependent on type I IFN signaling in mouse tracheal epithelial cultures and thus may represent a generalized response to viral infection [53••]. Similar to the studies with RSV, inhibition of PD-1 signaling during IAV infection enhances CD8+ T cell functions in a co-culture model and in the airways of infected mice [53••]. Thus, by increasing expression of PD-L1 by epithelial cells, viruses downregulate the effector functions of CD8+ T cells, thereby promoting viral replication.
22. GM-CSF is secreted by alveolar epithelial cells upon viral infection and also in response to TNF-α produced by stimulated alveolar macrophages [65••, 70]. GM-CSF promotes proliferation of AT2 cells, which leads to repair of damaged epithelium and restoration of its barrier functions [70].
23. type 1 alveoli epithelial cells produce more IL-1, IL-6, TNFalpha than AT2 cells in response to LPS stimulation. Exogenous surfactant decreases LPS-stimulated TI cell cytokine production
One of the most striking findings in this study was the dramatic increase in TNF-α and IL-6 production in the co-cultures of TI cells and macrophages. Exposing cultured TI cells to conditioned media from LPS-treated macrophages suggested that while TI cells do respond to inflammatory mediators liberated by activated macrophages by increasing TNF-α secretion, cell-cell interaction between macrophages and TI cells is a much more potent factor in modulating TI cell cytokine release. Conditioned media from activated macrophages decreased the levels of TI cell IL-6 compared to LPS-stimulated TI cells alone, while LPS treatment of co-cultures of TI cells and macrophages significantly increased IL-6 production. The IL-6 conditioned media experiments reinforce the notion that cell-cell interaction between TI cells and macrophages is powerful, as co-culture of these cells overcomes the inhibition of IL-6 production in TI cells by inhibitory mediators released by activated macrophages. While the exact cell of origin of the IL-6 in this instance is unclear, these results not only make a strong case for TI cells as key participants in the immune response of the lung, but they also suggest that the interaction between macrophages and TI cells may be equally important.
Our data has shown that TI and TII cells not only respond differently to LPS stimulation, but there exists the possibility that the two alveolar cell types work in concert within their microenvironment to provide a balanced inflammatory response to infection. One could postulate that TI cells may serve to heighten the pro-inflammatory response with enhanced cytokine production, particularly in the presence of alveolar macrophages, while TII cells, through the production of surfactant and relatively modest cytokine expression, provide more of an anti-inflammatory response by trying to contain inflammation.
https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0055545
http://cleverwaysoflearning.tumblr.com/
http://www.bio.miami.edu/tom/courses/bil265/bil265goods/15_hormones2.html
HIihtlights
1. H2O2 up-regulates Fas expression through the activation of protein tyrosine kinase in ECs.
2, Various cells express Fas, whereas Fas-L is expressed predominantly in activated T cells.
3. Vitamin C is an inhibitor of protein tyrosine kinase
Receptor Tyrosine Kinases (RTKs) in the Akt Pathway
Receptor Tyrosine Kinases (RTKs) are widely expressed transmembrane proteins that act as receptors for growth factors, neurotrophic factors, and other extracellular signaling molecules. Upon ligand binding, they undergo tyrosine phosphorylation at specific residues in the cytoplasmic tail. This leads to the binding of protein substrates and/or the establishment of docking sites for adaptor proteins involved in RTK-mediated signal transduction. The RTKs listed below have been associated with activation of the Akt signaling pathway, which promotes cell growth, survival, and proliferation. Due to these downstream effects, unregulated activation of RTKs can lead to cancer. Highlighting the significant role RTKs can have in cancer, small-molecule tyrosine kinase inhibitors are commonly used in cancer therapy.
Receptor Tyrosine Kinases (RTKs) in the Akt Pathway ...
http://www.cell.com/trends/pharmacological-sciences/fulltext/S0165-6147(11)00159-3
A tyrosine kinase is an enzyme that can transfer a phosphate group from ATP to a protein in a cell. It functions as an "on" or "off" switch in many cellular functions. Tyrosine kinases are a subclass of protein kinase. The phosphate group is attached to the amino acid tyrosine on the protein.en.wikipedia.org/wiki/Tyrosine_kinase
AECs mediate innate immunity
AECs act as a first line of defense against antigens that have escaped mucociliary clearance. Single cell‐RNA sequencing studies have shown that AECs can be subdivided into multiple subtypes 3, 5. The best characterized subtypes include squamous type I and cuboidal type II cells, which are responsible for gas exchange with the endothelium and production of pulmonary surfactant, respectively 11. Being endowed with an arsenal of pattern recognition receptors (PRRs) such as Toll‐like receptors (TLRs), Nod‐like receptors and retinoic acid‐inducible gene (RIG)‐I‐like receptors, AECs promptly sense pathogen‐associated molecules and initiate NF‐κB‐dependent inflammatory cascades through the release of antimicrobial peptides, cytokines and chemokines 11. Of particular importance are microbial ssRNA‐ and CpGDNA‐sensing TLR‐3, TLR‐7, TLR‐9, RIG‐I and melanoma differentiation‐associated protein‐5, which have been implicated in the rapid release of interferon (IFN)‐β, granulocyte‐macrophage colony‐stimulating factor (GM‐CSF), interleukin (IL)‐6 and IL‐8, a potent neutrophil‐recruiting chemokine 12. This robust cytokine response is critical to subsequently stimulate adaptive immunity, and neutralize the harmful effects of foreign pathogens.
However, owing to the hypersensitivity of AEC PRRs, these receptors can sometimes become diverted to mediate autoimmunity. For example, in mouse models of allergic asthma, activation of TLR‐4 by dust mites on AECs is associated with local production of IL‐25, IL‐33, GM‐CSF and thymic stromal lymphopoietin (TSLP) 13. These cytokines work in concert to stimulate activation and pulmonary infiltration of dendritic cells (DCs), lymphocytes, neutrophils and eosinophils 13. In addition, studies of pulmonary alveolar proteinosis have shown that GM‐CSF is specifically necessary for lung homeostasis, such that GM‐CSF autoantibodies or genetic deletion causes abnormal lung development and surfactant accumulation, despite normal peripheral hematopoiesis 14-16. Together these studies highlight a unique ability of AECs to undergo ‘innate immune mimicry’, and orchestrate inflammation and autoimmunity. Therefore, the contribution of AECs in the pulmonary microenvironment is essential to understanding both normal and pathologic lung function.The innate immune architecture of lung tumors and its implication in disease progression - Milette - 2019 - The Journal of Pathology - Wiley Online Library
https://onlinelibrary.wiley.com/doi/10.1002/path.5241
ijms-20-00831-g001.png (3272×2795)
https://www.mdpi.com/ijms/ijms-20-00831/article_deploy/html/images/ijms-20-00831-g001.png
F10: Diagram showing main findings of the paper. Using scanning electron microscopy a number of differences are visible in ALI/ARDS mouse lungs when compared to lungs of non-infected mice. In summary the endothelium of the capillaries ALI/ARDS lungs are swollen with distended cytoplasmic extensions and thickened basement membranes. The capillaries themselves are completely congested with leukocytes, niRBC and iRBC, in some instances bridges between iRBC and endothelium surfaces are visible. The alveolar space contains oedema, inflammatory cells and projections from septum epithelium, the septum are thick and full of leukocytes. ALI/ARDS: acute lung injury/acute respiratory distress syndrome. The layout of a single alveoli split to compare non-diseased and diseased lungs is based on a number of previous papers and books [25,16,27].
https://openi.nlm.nih.gov/detailedresult.php?img=PMC4062769_1475-2875-13-230-10&req=4
https://www.lhsc.on.ca/critical-care-trauma-centre/acute-respiratory-distress-syndrome-ards
J Virol. 2015 Apr;89(8):4655-67. doi: 10.1128/JVI.03095-14. Epub 2015 Feb 11.
A(H7N9) virus results in early induction of proinflammatory cytokine responses in both human lung epithelial and endothelial cells and shows increased human adaptation compared with avian H5N1 virus.
Zeng H1, Belser JA1, Goldsmith CS2, Gustin KM1, Veguilla V1, Katz JM1, Tumpey TM3.
Author information
1
Immunology and Pathogenesis Branch, Influenza Division, National Center for Immunization and Respiratory Diseases, Centers for Disease Control and Prevention, Atlanta, Georgia, USA.
2
Infectious Disease Pathology Branch, Division of High Consequence Pathogens and Pathology, National Center for Emerging and Zoonotic Infectious Diseases, Centers for Disease Control and Prevention, Atlanta, Georgia, USA.
3
Immunology and Pathogenesis Branch, Influenza Division, National Center for Immunization and Respiratory Diseases, Centers for Disease Control and Prevention, Atlanta, Georgia, USA tft9@cdc.gov.
Abstract
Similar to H5N1 viruses, A(H7N9) influenza viruses have been associated with severe respiratory disease and fatal outcomes in humans. While high viral load, hypercytokinemia, and pulmonary endothelial cell involvement are known to be hallmarks of H5N1 virus infection, the pathogenic mechanism of the A(H7N9) virus in humans is largely unknown. In this study, we assessed the ability of A(H7N9) virus to infect, replicate, and elicit innate immune responses in both human bronchial epithelial cells and pulmonary microvascular endothelial cells, compared with the abilities of seasonal H3N2, avian H7N9, and H5N1 viruses. In epithelial cells, A(H7N9) virus replicated efficiently but did not elicit robust induction of cytokines like that observed for H5N1 virus. In pulmonary endothelial cells, A(H7N9) virus efficiently initiated infection; however, no released infectious virus was detected. The magnitudes of induction of host cytokine responses were comparable between A(H7N9) and H5N1 virus infection. Additionally, we utilized differentiated human primary bronchial and tracheal epithelial cells to investigate cellular tropism using transmission electron microscopy and the impact of temperature on virus replication. Interestingly, A(H7N9) virus budded from the surfaces of both ciliated and mucin-secretory cells. Furthermore, A(H7N9) virus replicated to a significantly higher titer at 37 °C than at 33 °C, with improved replication capacity at 33 °C compared to that of H5N1 virus. These findings suggest that a high viral load from lung epithelial cells coupled with induction of host responses in endothelial cells may contribute to the severe pulmonary disease observed following H7N9 virus infection. Improved adaptation of A(H7N9) virus to human upper airway poses an important threat to public health.
IMPORTANCE:
A(H7N9) influenza viruses have caused over 450 documented human infections with a 30% fatality rate since early 2013. However, these novel viruses lack many molecular determinants previously identified with mammalian pathogenicity, necessitating a closer examination of how these viruses elicit host responses which could be detrimental. This study provides greater insight into the interaction of this virus with host lung epithelial cells and endothelial cells, which results in high viral load, epithelial cell death, and elevated immune response in the lungs, revealing the mechanism of pathogenesis and disease development among A(H7N9)-infected patients. In particular, we characterized the involvement of pulmonary endothelial cells, a cell type in the human lung accessible to influenza virus following damage of the epithelial monolayer, and its potential role in the development of severe pneumonia caused by A(H7N9) infection in humans.
Copyright © 2015, American Society for Microbiology. All Rights Reserved.A(H7N9) virus results in early induction of proinflammatory cytokine responses in both human lung epithelial and endothelial cells and shows increa... - PubMed - NCBI
https://www.ncbi.nlm.nih.gov/pubmed/25673714
HARVARD EALTH & MEDICINE
Study confirms vitamin D protects against colds and flu
A recent study by a global team of researchers has found that Vitamin D supplements, already widely prescribed for a variety of ailments, are effective in preventing respiratory diseases.
A new global collaborative study has confirmed that vitamin D supplementation can help protect against acute respiratory infections. The study, a participant data meta-analysis of 25 randomized controlled trials including more than 11,000 participants, has been published online in The BMJ.
“Most people understand that vitamin D is critical for bone and muscle health,” said Carlos Camargo of the Department of Emergency Medicine at Massachusetts General Hospital (MGH), the study’s senior author. “Our analysis has also found that it helps the body fight acute respiratory infection, which is responsible for millions of deaths globally each year.”
Several observational studies, which track participants over time without assigning a specific treatment, have associated low vitamin D levels with greater susceptibility to acute respiratory infections. A number of clinical trials have been conducted to investigate the protective ability of vitamin D supplementation, but while some found a protective effect, others did not. Meta-analyses of these trials, which aggregate data from several studies that may have different designs or participant qualifications, also had conflicting results.
To resolve these discrepancies, the research team — led by Adrian Martineau from Queen Mary University of London — conducted an individual participant data meta-analysis of trials in more than a dozen countries, including the U.S., Canada, and the U.K. While traditional meta-analyses compare average data from all participants in each study, individual participant data meta-analysis separates out the data from each individual participant, producing what could be considered a higher-resolution analysis of the data from all studies.
The investigators found that daily or weekly supplementation had the greatest benefit for individuals with the most significant vitamin D deficiency (blood levels below 10 mg/dl) — cutting their risk of respiratory infection in half — and that all participants experienced some beneficial effects from regular vitamin D supplementation. Administering occasional high doses of vitamin D did not produce significant benefits.
“Acute respiratory infections are responsible for millions of emergency department visits in the United States,” said Camargo, who is a professor of emergency medicine at Harvard Medical School. “These results could have a major impact on our health system and also support efforts to fortify foods with vitamin D, especially in populations with high levels of vitamin D deficiency.”
The study was funded by a grant from the National Institute of Health Research (U.K.).
哈佛医学院
研究证实维生素D可预防感冒和流感
由全球研究人员组成的团队最近进行的一项研究发现,维生素D补充剂已被广泛用于多种疾病,可有效预防呼吸道疾病。
一项全球合作研究表明,补充维生素D有助于预防急性呼吸道感染。该研究是一项包括25个随机对照试验的参与者数据荟萃分析,其中包括11,000多名参与者,已在线发表在BMJ上。
该研究的资深作者,麻省总医院(MGH)急诊科的卡洛斯·卡玛戈(Carlos Camargo)说:“大多数人都知道维生素D对骨骼和肌肉健康至关重要。” “我们的分析还发现,它可以帮助人体抵抗急性呼吸道感染,而这种疾病每年在全球造成数百万人死亡。”
几项观察性研究随时间推移追踪参与者而未分配具体治疗方法,这些研究发现维生素D含量低与急性呼吸道感染的易感性有关。已经进行了许多临床试验来研究补充维生素D的保健作用,但是尽管一些发现了保护作用,但其他人却没有。这些试验的荟萃分析汇总了一些研究的数据,这些研究可能具有不同的设计或参加者资格,但结果也不一致。为解决这些差异,由伦敦玛丽大学(Queen Mary University of London)的阿德里安·马丁诺(Adrian Martineau)领导的研究小组在包括美国,加拿大和英国在内的十几个国家/地区对试验的参与者数据进行了荟萃分析,而传统的分析会比较每个研究中所有参与者的平均数据,个体参与者数据荟萃分析会从每个个体参与者中分离出数据,从而可以对所有研究的数据进行更高分辨率的分析。
研究人员发现,每天或每周补充维生素D对维生素D缺乏症最严重的人(血液水平低于10 mg / dl)具有最大的益处-将其呼吸道感染的风险降低了一半-并且所有参与者都从常规补充维生素D中获得益处。偶尔服用大剂量维生素D并没有产生明显的益处。
哈佛医学院的急诊医学教授卡玛戈说:“急性呼吸道感染导致美国数百万急诊就诊。” “这些结果可能会对我们的卫生系统产生重大影响,并且还支持努力强化含维生素D的食品,特别是在维生素D缺乏水平高的人群中。”
该研究由英国国立卫生研究院(National Institute of Health Research)资助。Study confirms vitamin D protects against colds and flu – Harvard Gazette
https://news.harvard.edu/gazette/story/2017/02/study-confirms-vitamin-d-protects-against-cold-and-flu/
7.12: Virus Replication
Last updatedJun 15, 2019
7.11: Discovery and Origin of Viruses
7.13: Viruses and Human Disease
Notice the viruses sitting on the bacteria?
Why is the virus sitting here? Remember, viruses are not living. So how do they replicate?
Replication of Viruses
Populations of viruses do not grow through cell division because they are not cells. Instead, they use the machinery and metabolism of a host cell to produce new copies of themselves. After infecting a host cell, a virion uses the cell’s ribosomes, enzymes, ATP, and other components to replicate. Viruses vary in how they do this. For example:
Some RNA viruses are translated directly into viral proteins in ribosomes of the host cell. The host ribosomes treat the viral RNA as though it were the host’s own mRNA.
Some DNA viruses are first transcribed in the host cell into viral mRNA. Then the viral mRNA is translated by host cell ribosomes into viral proteins.
In either case, the newly made viral proteins assemble to form new virions. The virions may then direct the production of an enzyme that breaks down the host cell wall. This allows the virions to burst out of the cell. The host cell is destroyed in the process. The newly released virus particles are free to infect other cells of the host.
Replication of RNA Viruses
An RNA virus is a virus that has RNA as its genetic material. Their nucleic acid is usually single-stranded RNA, but may be double-stranded RNA. Important human pathogenic RNA viruses include the Severe Acute Respiratory Syndrome (SARS) virus, Influenza virus, and Hepatitis C virus. Animal RNA viruses can be placed into different groups depending on their type of replication.
Some RNA viruses have their genome used directly as if it were mRNA. The viral RNA is translated directly into new viral proteins after infection by the virus.
Some RNA viruses carry enzymes which allow their RNA genome to act as a template for the host cell to a form viral mRNA.
Retroviruses use DNA intermediates to replicate. Reverse transcriptase, a viral enzyme that comes from the virus itself, converts the viral RNA into a complementary strand of DNA, which is copied to produce a double stranded molecule of viral DNA. This viral DNA is then transcribed and translated by the host machinery, directing the formation of new virions. Normal transcription involves the synthesis of RNA from DNA; hence, reverse transcription is the reverse of this process. This is an exception to the central dogma of molecular biology.
Replication of DNA Viruses
A DNA virus is a virus that has DNA as its genetic material and replicates using a DNA-dependent DNA polymerase. The nucleic acid is usually double-stranded DNA but may also be single-stranded DNA. The DNA of DNA viruses is transcribed into mRNA by the host cell. The viral mRNA is then translated into viral proteins. These viral proteins then assemble to form new viral particles.
Reverse-Transcribing Viruses
A reverse-transcribing virus is any virus which replicates using reverse transcription, the formation of DNA from an RNA template. Some reverse-transcribing viruses have genomes made of single-stranded RNA and use a DNA intermediate to replicate. Others in this group have genomes that have double-stranded DNA and use an RNA intermediate during genome replication. The retroviruses, as mentioned above, are included in this group, of which HIV is a member. Some double-stranded DNA viruses replicate using reverse transcriptase. The hepatitis B virus is one of these viruses.
Bacteriophages are viruses that infect bacteria. They bind to surface receptor molecules of the bacterial cell and then their genome enters the cell. The protein coat does not enter the bacteria. Within a short amount of time, in some cases, just minutes, bacterial polymerase starts translating viral mRNA into protein. These proteins go on to become either new virions within the cell, helper proteins which help assembly of new virions, or proteins involved in cell lysis. Viral enzymes aid in the breakdown of the cell membrane. With some phages, just over twenty minutes after the phage infects the bacterium, over three hundred phages can be assembled and released from the host.
为什么新型冠状病毒在人体内复制这么快?
新型冠状病毒是一种RNA病毒。
RNA病毒复制
RNA病毒是一种以RNA为遗传物质的病毒。它们的核酸通常是单链RNA,但也可能是双链RNA。重要的人类致病性RNA病毒包括严重急性呼吸系统综合症(SARS)病毒和新型冠状病毒,流感病毒和丙型肝炎病毒。根据其复制类型,可以将动物RNA病毒分为不同的组。
有些RNA病毒的基因组直接用作mRNA。病毒感染后,病毒RNA可直接翻译成新的病毒蛋白。
一些RNA病毒携带的酶允许其RNA基因组充当宿主细胞形成病毒mRNA的模板。
逆转录病毒使用DNA中间体复制。逆转录酶是一种来自病毒本身的病毒酶,可将病毒RNA转化为DNA的互补链,将其复制以产生病毒DNA的双链分子。然后,该病毒DNA被宿主机器转录并翻译,指导新病毒体的形成。正常转录涉及从DNA合成RNA。因此,逆转录是该过程的逆过程。这是分子生物学中心教条的一个例外。7.12: Virus Replication - Biology LibreTexts
https://bio.libretexts.org/Bookshelves/Introductory_and_General_Biology/Book%3A_Introductory_Biology_(CK-12)/7%3A_Prokaryotes_and_Viruses/7.12%3A_Virus_Replication
Lopinavir罗匹那韦
蛋白酶抑制剂,抗逆转录病毒。
洛匹那韦的作用机理
罗匹那韦是肽类似物。它可逆地结合到HIV编码的天冬氨酰蛋白酶的催化位点,该酶参与多蛋白降解成结构蛋白和随后的病毒颗粒成熟。洛匹那韦与酶的结合导致非感染性病毒后代。
洛匹那韦的药代动力学
吸收:口服吸收
分布:主要以蛋白质结合形式分布
代谢:它在肝脏中进行新陈代谢。
排泄:药物及其代谢物主要通过粪便排出。Ritonavir
About Ritonavir
HIV Protease Inhibitor, Thiazole derivative, Antiretroviral.
Mechanism of Action of Ritonavir
Ritonavir is a peptide analogue. It reversibly binds to the catalytic site of HIV encoded aspartyl protease enzyme which is involved in the degradation of poly protein into structural protein and subsequent maturation of virus particle. Binding of Ritonavir to the enzyme results in immature noninfectious viral progeny
Pharmacokinets of Ritonavir
Absorption: It is rapidly absorbed after oral administration. Distribution: It is distributed as protein bound form. Metabolism: It undergoes metabolism in the liver. Excretion: It is excreted primarily through faeces and small amount through urineRitonavir利托那韦
HIV蛋白酶抑制剂,噻唑衍生物,抗逆转录病毒。
利托那韦的作用机理
利托那韦是肽类似物。它可逆地结合到HIV编码的天冬氨酰蛋白酶的催化位点,该酶参与多蛋白降解成结构蛋白和随后的病毒颗粒成熟。利托那韦与酶的结合导致非感染性病毒后代
利托那韦的药代动力学
吸收:口服后迅速吸收。分布:它以蛋白质结合形式分布。代谢:它在肝脏中进行新陈代谢。排泄:主要通过粪便排出,少量通过尿排出
新型冠状病毒感染肺炎疫情时期的合理营养与膳食建议
发布时间:2020-02-03
张立实
四川大学华西公共卫生学院 教授/博士生导师
中国营养学会常务理事/四川省营养学会理事长
大家都已经知道,对于新型冠状病毒感染肺炎,目前还没有特异性的疫苗和治疗药物。这种传染性疾病的发生、发展与预后(死亡或康复),很大程度上取决于个体的免疫功能及其营养、健康状况(如:是否有营养不良或营养失衡,是否患有慢性非传染性疾病,体质情况等)。因此,通过合理膳食来增强机体的免疫功能和身体素质,提高抗病能力,对预防新型冠状病毒感染肺炎的发生或减轻其病情有非常重要的意义。
在此,对新型冠状病毒感染肺炎疫情时期一般人群(不包括新型冠状病毒感染肺炎患者和疑似患者、疫情一线的医务工作者和其他人员,以及其他疾病患者等)的合理营养与膳食给出以下几点简明扼要的建议:
1. 重视食物多样化,尽量做到膳食平衡
每天尽可能在各大类食物(谷薯类、蔬菜水果类、奶蛋类、禽畜肉和鱼虾类、大豆坚果类)中选择多种食物,以满足人体基本的营养需求和平衡膳食的需要。同时,由于活动和运动减少,应适当控制总能量和高脂肪食物(肥肉、动植物油脂、油炸食品等)的摄入,以“食至七、八分饱”和维持健康体重为宜。
2. 保证蛋白质,尤其是优质蛋白质的足量摄入
蛋白质是构成人体组织细胞的主要成分,具有多方面的重要生理作用,尤其对于维持机体的正常免疫功能、防御病毒感染有至关重要的作用。因此,要保证每天摄入充足的蛋白质,尤其是优质蛋白质。富含优质蛋白质的食物包括瘦肉、鱼、虾、蛋、奶和大豆(制品)等,每日摄入瘦肉和鱼虾类应不少于100g,并尽量保证每天吃一个鸡蛋;奶类、大豆(制品)和坚果类等也可适当多选择食用。
3. 适量多吃新鲜蔬菜和水果
新鲜蔬菜水果中富含维生素类(尤其是维生素C)、矿物质和微量元素、有机酸以及多种多样的植物化学物(如黄酮类、多酚类、多糖类、花青素和类胡萝卜素、含硫化合物、皂苷类等),具有不同程度的增强免疫力和抗氧化等作用,对防御病毒感染有一定益处。建议每天选择食用至少4~5种蔬菜(其中深色蔬菜不少于一半),2~3种水果,蔬果总量应不少于500克。如因食物供应和购买方面等客观原因不能充分保证新鲜蔬果的种类和数量,可选择适量的干菜干果加以补充。菌菇类富含香菇多糖等具有增强免疫功能的化合物,可适量多吃。大蒜、洋葱、葱等辛辣味重的蔬菜含有较多的含硫化合物,可提高体内免疫细胞的活力,也有预防病毒感染的作用,亦可适量食用。
4. 重视食品安全
非常时期尤其要重视食品安全,避免因食品卫生问题而发生食物中毒和其他食源性疾病;不要购买和食用野生动物,以及某些平常没有吃过或很少食用的食物;食物准备和烹饪、制作者更要注意个人卫生,勤洗手,并保证“生熟分开”操作;尽量少吃生冷食物,尤其动物性食物要烧熟煮透再吃;尽量不要外出聚餐,家庭用餐也建议使用分餐制或使用公勺公筷等,尤其对于“在家隔离”的情况,更要十分重视分开就餐以及食物和就餐环境的卫生。
5. 保证充足的饮水量
足量的水是维持人体内物质代谢和正常生理功能(包括免疫功能)所必需,故应保证每日有充足的饮水量。不同个体的水代谢和饮水习惯常有较大差异,应在平常饮水量的基础上略有增加,一般每天应不少于2000ml(约5大茶杯),并建议多饮白开水或茶水,尽量少喝高糖饮料和碳酸饮料。
6.适当补充维生素和保健食品
在食物供应和种类、数量有限,或特殊健康原因(如免疫力低下、微量营养素缺乏或需要量增加)等情况下,可选择服用复合维生素矿物质补充剂和具有增强免疫力功能的保健食品。
7. 保持适当的运动/活动量和良好的心态
因疫情防控需要而尽可能少外出,更多时间是“宅家”,在这种情况下,更应重视保持良好的心态,相信科学,相信党和政府,对战胜疫情要有足够的信心。同时,要注意每天保持一定的运动/活动量,视不同的居家环境条件和个人爱好,进行适当的室内运动/活动;在条件允许的情况下,也可到空旷人少的地方(如公园、校园等)进行适当的户外活动。https://www.cnsoc.org/learnnews/322000201.html
Human Beta Defensin 2 Selectively Inhibits HIV-1 in Highly Permissive CCR6+CD4+ T Cells
by Mark K. Lafferty 1,2, Lingling Sun 1, Aaron Christensen-Quick 1,2, Wuyuan Lu 1,3 and Alfredo Garzino-Demo 1,2,4,*OrcID
1
Division of Basic Science, Institute of Human Virology, University of Maryland School of Medicine, Baltimore, MD 21201, USA
2
Department of Microbiology and Immunology, University of Maryland School of Medicine, Baltimore, MD 21201, USA
3
Department of Biochemistry, University of Maryland School of Medicine, Baltimore, MD 21201, USA
4
Department of Molecular Medicine, University of Padova, Padova 35121, Italy
*
Author to whom correspondence should be addressed.
Academic Editor: Theresa Chang
Viruses 2017, 9(5), 111; https://doi.org/10.3390/v9050111
Abstract
Chemokine receptor type 6 (CCR6)+CD4+ T cells are preferentially infected and depleted during HIV disease progression, but are preserved in non-progressors. CCR6 is expressed on a heterogeneous population of memory CD4+ T cells that are critical to mucosal immunity. Preferential infection of these cells is associated, in part, with high surface expression of CCR5, CXCR4, and α4β7. In addition, CCR6+CD4+ T cells harbor elevated levels of integrated viral DNA and high levels of proliferation markers.
We have previously shown that the CCR6 ligands MIP-3α and human beta defensins inhibit HIV replication. The inhibition required CCR6 and the induction of APOBEC3G. Here, we further characterize the induction of apolipoprotein B mRNA editing enzyme (APOBEC3G) by human beta defensin 2. Human beta defensin 2 rapidly induces transcriptional induction of APOBEC3G that involves extracellular signal-regulated kinases 1/2 (ERK1/2) activation and the transcription factors NFATc2, NFATc1, and IRF4.
We demonstrate that human beta defensin 2 selectively protects primary CCR6+CD4+ T cells infected with HIV-1. The selective protection of CCR6+CD4+ T cell subsets may be critical in maintaining mucosal immune function and preventing disease progression. View Full-Text
人类β防御素2选择性抑制高度允许的CCR6 + CD4 + T细胞中的HIV-1。抽象
6型趋化因子受体(CCR6)+ CD4 + T细胞在HIV疾病发展过程中会优先受到感染和消耗,但保留在非进展者中。 CCR6在对黏膜免疫至关重要的记忆CD4 + T细胞异质群体中表达。这些细胞的优先感染部分与CCR5,CXCR4和α4β7的高表面表达有关。此外,CCR6 + CD4 + T细胞具有较高水平的整合病毒DNA和高水平的增殖标记物。 先前我们已经表明,CCR6配体MIP-3α和人β防御素可抑制HIV复制。抑制作用需要CCR6和APOBEC3G的诱导。在这里,我们进一步表征了人类β防御素2对载脂蛋白B mRNA编辑酶(APOBEC3G)的诱导。人类β防御素2快速诱导APOBEC3G的转录诱导,涉及细胞外信号调节激酶1/2(ERK1 / 2)激活和转录因子NFATc2,NFATc1和IRF4。我们证明了人类β防御素2选择性保护感染了HIV-1的初级CCR6 + CD4 + T细胞。 CCR6 + CD4 + T细胞亚群的选择性保护对于维持粘膜免疫功能和预防疾病进展可能至关重要。
Viruses | Free Full-Text | Human Beta Defensin 2 Selectively Inhibits HIV-1 in Highly Permissive CCR6+CD4+ T Cells
https://www.mdpi.com/1999-4915/9/5/111#
Human Beta Defensins and Cancer: Contradictions and Common ...
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6509205
May 03, 2019 · The discovery of human β-defensins (hBDs) in mucosa has led to recognition that they are integral in innate immune protection; shielding mucosal surfaces from microbial challenges.
Cited by: 2
Publish Year: 2019
Author: Santosh K. Ghosh, Thomas S. McCormick, Aaron Weinberg
Fusobacterium nucleatum and Human Beta-Defensins Modulate ...
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3257922
Cells of the innate immune system regulate immune responses through the production of antimicrobial peptides, chemokines, and cytokines, including human beta-defensins (hBDs) and CCL20. In this study, we examined the kinetics of primary human oral epithelial ...
Cited by: 12
Publish Year: 2011
Author: Santosh K. Ghosh, Sanhita Gupta, Bin Jiang, Aaron Weinberg
Production of β-defensins by human airway epithelia | PNAS
https://www.pnas.org/content/95/25/14961.full
Furthermore, it is not known whether β-defensin production is deficient in diseases such as cystic fibrosis (CF) that are characterized by chronic infection. Recent studies have identified β-defensin expression at mucosal surfaces in human tissues, where they may play a role in innate defenses …
Cited by: 736
Publish Year: 1998
Author: Pradeep K. Singh, Hong Peng Jia, Kerry Wiles, Jay Hesselb
Effect of Human Beta Defensin-2 in Epithelial Cell Lines Infected with Respiratory Viruses
Abstract
Miguel Ángel Galván Morales, Alejandro Escobar Gutiérrez, Dora Patricia Rosete Olvera and Carlos Cabello Gutiérrez
β-defensins are a family of antimicrobial molecules involved in inflammatory processes and infections. In human airways, β-defensin-2 (hβD-2) is the best characterized in bacterial and fungal infections; however, it has been insufficiently studied in viral infections. The respiratory syncytial virus (RSV) and adenoviruses (ADV) are important agents of acute respiratory infections. The aim of this study was to measure in vitro the production and antiviral activity of hβD-2 in HEp-2 cells and A549 cells infected with ADV and RSV; hβD-2 production at different times was assessed by RT-PCR, and its presence by immunodetection assay (Western blot) using antibodies anti-hβD-2. The effect of this defensin on viral replication was determined using recombinant hβD-2 in plaque assays. The results revealed that in the cell lines production of hβD-2is up regulated after ADV or RSV infection, in direct proportion to the exposure time to each virus. The use of a high concentration of recombinant hβD-2 resulted in less deleterious viral effect on the cells. The results suggest that both viruses induce hβD-2 production, no matter if the virus is enveloped or not, and that presence of hβD-2 reduces replication and cytopathic in vitro effect of RSV and ADV. The hβD-2 production by low pathogenicity viruses or live viral vaccines can be useful as therapeutic tools in some infectious diseases.
人β-防御素-2 (Defensin-2)在呼吸道病毒感染的上皮细胞系中的作用
摘要
MiguelÁngelGalvánMorales,Alejandro EscobarGutiérrez,Dora Patricia Rosete Olvera和Carlos CabelloGutiérrez
β-防御素是涉及炎症过程和感染的一系列抗菌分子。在人的气道中,β-防御素-2(hβD-2)在细菌和真菌感染中表现最出色。但是,在病毒感染方面尚未进行充分的研究。呼吸道合胞病毒(RSV)和腺病毒(ADV)是急性呼吸道感染的重要媒介。这项研究的目的是在体外测量被ADV和RSV感染的HEp-2(人类上皮)细胞和A549细胞中hβD-2的产生和抗病毒活性。通过RT-PCR评估hβD-2在不同时间的产生,并使用抗hβD-2抗体通过免疫检测测定法(Western blot)评估其存在。在噬菌斑试验中使用重组hβD-2确定了这种防御素对病毒复制的作用。
结果表明,在ADV或RSV感染后,hβD-2的生产受到上调,与每种病毒的暴露时间成正比。高浓度重组hβD-2的使用对病毒的细胞有害作用较小。
结果表明,无论是否被包膜病毒,两种病毒均诱导hβD-2的产生,并且hβD-2的存在会降低RSV和ADV的复制和体外细胞病变作用。低致病性病毒或活病毒疫苗产生的hβD-2可用作某些传染病的治疗工具。
Effect of Human Beta Defensin-2 in Epithelial Cell Lines Infected with Respiratory Viruses | Abstract
https://www.hilarispublisher.com/abstract/effect-of-human-beta-defensin2-in-epithelial-cell-lines-infected-with-respiratory-viruses-28005.html
Int J Biochem Cell Biol. 1999 Jun;31(6):645-51.
Human beta-defensin-2.
Schröder JM1, Harder J.
1 Department of Dermatology, University of Kiel, Germany. jschroeder@dermatology.uni-kiel.de
Abstract
Human beta-defensin-2 (HBD-2) is a cysteine-rich cationic low molecular weight antimicrobial peptide recently discovered in psoriatic lesional skin. It is produced by a number of epithelial cells and exhibits potent antimicrobial activity against Gram-negative bacteria and Candida, but not Gram-positive Staphylococcus aureus. HBD-2 represents the first human defensin that is produced following stimulation of epithelial cells by contact with microorganisms such as Pseudomonas aeruginosa or cytokines such as TNF-alpha and IL-1 beta. The HBD-2 gene and protein are locally expressed in keratinocytes associated with inflammatory skin lesions such as psoriasis as well as in the infected lung epithelia of patients with cystic fibrosis. It is intriguing to speculate that HBD-2 is a dynamic component of the local epithelial defense system of the skin and respiratory tract having a role to protect surfaces from infection, and providing a possible reason why skin and lung infections with Gram-negative bacteria are rather rare.
PMID: 10404637 DOI: 10.1016/s1357-2725(99)00013-8人β-防御素2(HBD-2)是一种最近在银屑病皮损皮肤中发现的富含半胱氨酸的阳离子低分子量抗菌肽。它由许多上皮细胞产生,对革兰氏阴性菌和念珠菌具有有效的抗菌活性,但对革兰氏阳性金黄色葡萄球菌则没有。 HBD-2代表第一个人类防御素,是通过与铜绿假单胞菌等微生物或TNF-α和IL-1β等细胞因子接触刺激上皮细胞而产生的。 HBD-2基因和蛋白在与炎症性皮肤病(如牛皮癣)相关的角质形成细胞中以及在患有囊性纤维化患者的被感染的肺上皮中局部表达。有趣的是,HBD-2是皮肤和呼吸道局部上皮防御系统的动态成分,具有保护表面不受感染的作用,是皮肤和肺部革兰氏阴性细菌感染相当罕见的可能原因。
Human beta-defensin-2. - PubMed - NCBI
https://www.ncbi.nlm.nih.gov/pubmed/10404637
Human beta-defensin 2 is a salt-sensitive peptide antibiotic expressed in human lung.
R Bals, X Wang, Z Wu, T Freeman, V Bafna, M Zasloff, and J M Wilson
First published September 1, 1998 - More info
Institute for Human Gene Therapy, Department of Medicine and Molecular and Cellular Engineering, The Wistar Institute, Philadelphia, Pennsylvania 19104, USA.
Abstract
Previous studies have implicated the novel peptide antibiotic human beta-defensin 1 (hBD-1) in the pathogenesis of cystic fibrosis. We describe in this report the isolation and characterization of the second member of this defensin family, human beta-defensin 2 (hBD-2). A cDNA for hBD-2 was identified by homology to hBD-1. hBD-2 is expressed diffusely throughout epithelia of many organs, including the lung, where it is found in the surface epithelia and serous cells of the submucosal glands. A specific antibody made of recombinant peptide detected hBD-2 in airway surface fluid of human lung. The fully processed peptide has broad antibacterial activity against many organisms, which is salt sensitive and synergistic with lysozyme and lactoferrin. These data suggest the existence of a family of beta-defensin molecules on mucosal surfaces that in the aggregate contributes to normal host defense.
人β-防御素2是在人肺中表达的盐敏感性肽抗生素。
R Bals,X Wang,Z Z Wu,T Freeman,V Bafna,M Zasloff和J M Wilson
1998年9月1日首次发布-更多信息
美国宾夕法尼亚州费城,维斯塔研究所,医学与分子与细胞工程系,人类基因治疗研究所。
抽象
先前的研究已将新型肽类抗生素人β-防御素1(hBD-1)牵涉到囊性纤维化的发病机理中。我们在这份报告中描述了该防御素家族的第二个成员,人β-防御素2(hBD-2)的分离和鉴定。通过与hBD-1的同源性鉴定了hBD-2的cDNA。hBD-2在包括肺在内的许多器官的上皮中广泛表达,在粘膜下腺的表面上皮和浆液细胞中发现。由重组肽制成的特异性抗体在人肺气道表面液中检测到hBD-2。经过充分加工的肽对多种生物具有广泛的抗菌活性,对盐敏感并与溶菌酶和乳铁蛋白协同作用。这些数据表明,在粘膜表面上存在着一系列的β-防御素分子,这些分子总体上有助于正常的宿主防御。
JCI - Human beta-defensin 2 is a salt-sensitive peptide antibiotic expressed in human lung.
https://www.jci.org/articles/view/2410
Respir Res. 2005; 6(1): 116.
Respiratory epithelial cells require Toll-like receptor 4 for induction of Human β-defensin 2 by Lipopolysaccharide
Ruth MacRedmond,corresponding author1 Catherine Greene,1 Clifford C Taggart,1 Noel McElvaney,1 and Shane O'Neill1
Abstract
Background
The respiratory epithelium is a major portal of entry for pathogens and employs innate defense mechanisms to prevent colonization and infection. Induced expression of human β-defensin 2 (HBD2) represents a direct response by the epithelium to potential infection. Here we provide evidence for the critical role of Toll-like receptor 4 (TLR4) in lipopolysaccharide (LPS)-induced HBD2 expression by human A549 epithelial cells.
Methods
Using RTPCR, fluorescence microscopy, ELISA and luciferase reporter gene assays we quantified interleukin-8, TLR4 and HBD2 expression in unstimulated or agonist-treated A549 and/or HEK293 cells. We also assessed the effect of over expressing wild type and/or mutant TLR4, MyD88 and/or Mal transgenes on LPS-induced HBD2 expression in these cells.
Results
We demonstrate that A549 cells express TLR4 on their surface and respond directly to Pseudomonas LPS with increased HBD2 gene and protein expression. These effects are blocked by a TLR4 neutralizing antibody or functionally inactive TLR4, MyD88 and/or Mal transgenes. We further implicate TLR4 in LPS-induced HBD2 production by demonstrating HBD2 expression in LPS non-responsive HEK293 cells transfected with a TLR4 expression plasmid.
Conclusion
This data defines an additional role for TLR4 in the host defense in the lung.
Keywords: Airway epithelium, Toll-like Receptor 4, Lipopolysaccharide, Human β-defensin 2.
Introduction
The lung represents the largest epithelial surface in the body and is a major portal of entry for pathogenic microorganisms. It employs a number of efficient defense mechanisms to eliminate airborne pathogens encountered in breathing, including the specific innate and adaptive immune responses, which represent a dynamic interaction of host and pathogen. Lipopolysaccharide (LPS) is an important antigenic component of Gram-negative bacteria, and is a potent stimulus to local and systemic immune responses. The human receptor for LPS is Toll-like-receptor 4 (TLR4) [1].
TLRs are a family of pattern recognition receptors whose pivotal importance in orchestrating the innate immune response is widely accepted. Binding of ligand activates a signaling cascade involving TRAF6, IKKs and I-κBs, culminating in NF-κB translocation to the nucleus [1]. NF-κB regulates the inducible expression of cytokines, chemokines, adhesion molecules and acute phase proteins which activate cellular immune responses [2]. TLR signaling pathways arise from intracytoplasmic Toll/IL-1 receptor (TIR) domains, which are conserved among TLRs and TIR domain-containing adaptor proteins such as MyD88, Mal/TIRAP and TRIF/TICAM-1. These adaptor proteins confer specificity on TLR signaling, with Mal specifically involved in MyD88-dependent signaling via TLR2 and TLR4, and TRIF in the MyD88-independent TLR3- and TLR4- signaling [3]
The mammalian innate immune system produces a variety of anti-microbial peptides (AMPs) as part of its host defense repertoire. The defensins are a broadly dispersed group of AMPs, and are classified according to their molecular structure into three distinct families: the α-, β- and the θ-defensins. Unlike α-defensins, which are produced mainly by neutrophils, β-defensins are produced directly by epithelial cells, and combat infection both through direct microbicidal action and by modulation of cell-mediated immunity [4-7]. To date, four human β-defensins (HBD) have been identified (HBD1-4), although genomic studies suggest more have yet to be discovered [8,9]. In contrast to HBD1, which is constitutively and stably expressed, HBD2 expression is induced in response to infective stimuli, including Gram-negative and, less potently, Gram-positive bacteria or their components or to proinflammatory stimuli including tumor necrosis factor α (TNFα) and interleukin-1β (IL-1β) in vitro [10,11].
Like other defensins, HBD2 has a broad spectrum of antimicrobial activity, displaying potent microbicidal activity against many Gram-negative bacteria and less potent bacteriostatic activity against Gram-positive bacteria [11]. It has recently been demonstrated that activation of TLR2 by bacterial lipoprotein results in up regulation of HBD2 in tracheobronchial epithelium [12]. LPS and Gram-negative bacteria such as mucoid P. aeruginosa are a more potent stimulus for HBD2 production, which in turn has anti-bacterial activity predominantly against Gram-negative bacteria. Colonisation and infection due to Gram-negative bacteria are important in many pulmonary diseases including severe COPD [13] and Cystic Fibrosis [14]. Production of HBD2 by respiratory epithelium is an important component of host defense against Gram-negative organisms, and understanding of the signaling pathways involved may further our understanding of and guide future therapeutic strategies in these diseases.
Cultured intestinal epithelial cells have been shown to produce HBD2 in response to LPS following transfection with TLR4 and MD2 [15]. Although CD-14 is known to be critical to LPS-induced HBD2 production in airway epithelium [16], the role of TLR4 in transcriptional regulation of HBD2 in respiratory epithelium has not been established. Indeed, the importance of the respiratory epithelium in the innate immune response to LPS has been called into question by some recent publications [17,18]. In this study we demonstrate TLR4 expression in A549 pulmonary epithelial cells and production of HBD2 in response to LPS. We examine the effect of modulation of TLR4 by receptor blockade and expression of a dominant negative TLR4 construct on induced expression of HBD2. We show that LPS-unresponsive HEK293 cells can produce HBD2 in response to LPS following transfection with TLR4 and MD2 transgenes and demonstrate that the adaptor proteins MyD88 and Mal are involved in transcriptional regulation of HBD2 in response to LPS.Respiratory epithelial cells require Toll-like receptor 4 for induction of Human β-defensin 2 by Lipopolysaccharide
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1276817/
《浙江大学学报(医学版)》 2006年06期
重组β-防御素2多肽对脓毒症大鼠肺组织细胞凋亡的影响
徐笑益 石卓 鲍军明 顾伟忠 姚航平 陈智 高敏 方向明
【摘要】:目的:观察重组β-防御素2多肽预处理对脓毒症大鼠肺组织细胞凋亡的影响。方法:SPF级SD大鼠48只随机分成对照组和防御素组,采用盲肠结扎穿孔术(CLP)复制脓毒症模型,防御素组在CLP前48h经气管插管气道滴注107PFU重组腺病毒(含有β-防御素2编码基因),而对照组则同法给予对照腺病毒(不含β-防御素2编码基因)。分别于CLP后0、12、36和72 h处死大鼠,取肺组织,采用透射电镜,末端脱氧核苷酸转移酶介导的dUTP缺口末端标记法(TUNEL)检测细胞凋亡,采用电镜、苏木精-伊红(HE)染色观察肺组织病理变化。结果:TUNEL检测显示,对照组大鼠CLP后12、36和72 h肺组织细胞凋亡指数显著增加(P0.01);与对照组相比,防御素组CLP后相应时间点肺组织细胞凋亡指数显著降低(P0.05)。HE染色可见,两组大鼠CLP后12、36和72 h肺泡及肺泡间质充血、水肿,炎症细胞渗出,肺泡腔不同程度狭窄。与对照组相比较,防御素组相应时间点肺组织内炎症细胞渗出减少,间质水肿减轻。结论:重组β-防御素2多肽预处理能抑制脓毒症肺组织的细胞凋亡,对急性肺损伤(AL I)具有保护作用。
【作者单位】: 浙江大学医学院附属邵逸夫医院麻醉科 浙江大学医学院附属儿童医院儿科研究所 浙江大学医学院附属邵逸夫医院麻醉科 浙江大学医学院附属儿童医院儿科研究所 浙江大学医学院附属第一医院传染病研究所 浙江大学医学院附属第一医院传染病研究所 浙江大学医学院附属邵逸夫医院临床实验室 浙江大学医学院附属第一医院麻醉科
【基金】:国家自然科学基金资助项目(30070854)
【分类号】:R459.7重组β-防御素-2对呼吸道绿脓杆菌感染大鼠急性肺损伤的保护作用
王海宏 舒强 石卓 赵正言 方向明 【摘要】:目的 观察重组β-防御素-2对呼吸道绿脓杆菌感染大鼠急性肺损伤(ALI)的保护作用。 方法10只清洁级雄性成年SD大鼠,随机分为防御素组和对照组,每组5只。防御素组大鼠暴露声 门后,气管内滴注5×107PFU/ml重组腺病毒(含有β-防御素-2编码基因)50μl,对照组给予等量对照腺 病毒(不含β-防御素-2编码基因)。48 h后两组气管内滴注6×108CFU/ml绿脓杆菌ATCC27853 200μl, 制备绿脓杆菌感染致ALI模型。气管内滴注绿脓杆菌24 h后处死大鼠,采集肺泡灌洗液,进行绿脓杆 菌菌落数和白细胞计数;观察肺组织病理学变化,并测定肺组织细胞间粘附分子-1(ICAM-1)表达水 平。结果与对照组比较,防御素组BALF中绿脓杆菌菌落数、白细胞计数、肺组织ICAM-1表达水平 及肺病理组织学评分降低(P0.05)。结论重组β-防御素-2对呼吸道绿脓杆菌感染大鼠ALI有一 定的保护作用,可能与其杀菌作用和下调肺组织ICAM-1的表达有关。 【作者单位】: 【基金】:国家自然科学基金资助项目(30070854) 【分类号】:R96
《中国呼吸与危重监护杂志》 2010年01期
β-防御素2与下呼吸道感染患者全身炎症反应的关系
刘松 何丽蓉 贺正一 王浩彦
【摘要】:目的研究下呼吸道感染(LRTI)患者外周血β-防御素2(HBD-2)与全身炎症反应的关系。方法LRTI组纳入81例患者,包括社区获得性肺炎、COPD急性加重或(和)合并肺部感染、支气管扩张合并感染。同期20例健康人作为对照组。酶联免疫吸附法(ELISA)测定外周血HBD-2、白细胞介素1β(IL-1β)和IL-8水平。常规进行血细胞分析等实验室检查。比较LRTI组及对照组上述指标的变化。按照采血时患病时间分为7d组、7~14d组和14d组,比较组间差别。对HBD-2与IL-1β、IL-8进行相关分析。结果LRTI组患者外周血HBD-2、IL-1β和白细胞总数(WBC)显著高于对照组(P均0.05)。LRTI患者在患病时间7d组和7~14d组的HBD-2和IL-1β水平均显著高于对照组(P均0.05),14d组患者的HBD-2和IL-1β基本恢复正常(P均0.05)。不同患病时间的LRTI患者IL-8水平无显著差异(P均0.05)。外周血HBD-2与IL-1β呈显著正相关(r=0.313,P=0.030);HBD-2与IL-8无显著相关性(P0.05)。结论LRTI患者外周血HBD-2明显升高,HBD-2升高主要表现在疾病早期或相对早期。外周血HBD-2升高可能与全身炎症反应有关。
【作者单位】: 首都医科大学附属北京友谊医院呼吸内科;
In this study, bovine lactoferrin (bLF) was used in both in vitro and in vivo approaches to investigate its activity against lung cancer. A human lung cancer cell line, A549, which expresses a high level of vascular endothelial growth factor (VEGF) under hypoxia, was used as an in vitro system for bLF treatment.
Bovine lactoferrin inhibits lung cancer growth through ...
www.sciencedirect.com/science/article/pii/S0022030213001434
Tips for healthy respiratory system
People often take healthy respiratory system for granted as breathing happens almost automatically. However, by taking proper measures one can increase the health of the respiratory system.
LUNG DISEASES By : Meenakshi Chaudhary , Onlymyhealth Editorial Team / Date : Jan 25, 2014
RESPIRATORY SYSTEM
People often take healthy respiratory system for granted as breathing happens almost automatically. But the fact that even a slight decrease in the lung function cannot be ignored as it can cause disorder to the rest of the body declining your overall health. However, by taking proper measures one can increase the health of the respiratory system.
EAT HEALTHY
Healthy eating is important to maintain the health of your lungs. Our body needs vitamins and nutrients to work effectively and build the tissues including the ones that make up the respiratory system. According to some studies, healthy fats such as omega-3 fatty acids help in inhibiting the inflammation in the lungs and provide an ameliorative effect to the asthma patients.
QUIT SMOKING
If you are a smoker, one of the most important changes that can be made to improve the respiratory health is stop smoking. Tar and thousands of chemicals of the cigarette smoke tend to reduce the capacity and efficiency of your lungs. Long term smoking can result in irreversible chronic respiratory problems. Quitting smoking not only helps in reversing the acute health problems but helps in improving the respiratory health.
EXERCISE
Regular aerobic exercise can help you improve the respiratory health. Exercise increases need of oxygen to your muscles causing the brain to stimulate the respiratory system to increase ventilation or breathing. This increase in the frequency and depth of breathing expands your lungs, enhances the elasticity of the air sacs, expels old air within your lungs and tones and strengthens the diaphragm. All these changes have a lasting effect which helps in improving respiratory health even after you have stopped exercising.
OBESITY
Obesity is a result of bad eating and lifestyle habits which may have bad effects on your respiratory system. A low cholesterol diet is good for your overall health while a diet rich in refined sugars and trans fats will contribute to poor respiratory system.
AVOID POLLUTION
Pollution and urban smog can cause respiratory problems like asthma, bronchitis and lung cancer. If you live in a large city, wearing a face mask will act as a protective barrier to your respiratory system.
FAST FOOD
Fast food can increase your chances of having a respiratory disease. Switch to a healthy diet for a strong respiratory system. Food like dairy may also harm your respiration. Consuming dairy can trigger asthma attacks and contribute to the general poor health of the respiratory system. So keep a check on your fast food and dairy intake.
SLEEP
Several studies have shown that people who get an extra hour of sleep at night have a lower risk for respiratory problems and artery-clogging calcification that can lead to heart disease. People who don't get enough sleep are more prone to respiratory problems.
ALCOHOL
Alcohol consumption can lead to high blood pressure which can lead to respiratory problems and heart failure. Anything more than moderate intake of alcohol may be harmful to your overall health.
AVOID ALLERGENS
Common allergens like dust mites, pollen, mold, and animal dander can be harmful for your respiratory system. Vacuum and damp dust all surfaces regularly. Ensure that your ac filters, curtains and carpet is regularly cleaned with similar chemical solutions.
促进呼吸系统健康的提示
人们常常认为健康的呼吸系统是理所当然的,因为呼吸几乎是自动发生的。但是,通过采取适当的措施,可以提高呼吸系统的健康。
肺部疾病作者:Meenakshi Chaudhary,Onlymyhealth编辑部团队/日期:2014年1月25日
呼吸系统
人们常常认为健康的呼吸系统是理所当然的,因为呼吸几乎是自动发生的。但是,即使肺功能略有下降,这一事实也不容忽视,因为它可能导致身体其他部位的不适,从而影响您的整体健康。但是,通过采取适当的措施,可以提高呼吸系统的健康。
吃得健康
健康饮食对维持肺部健康至关重要。我们的身体需要维生素和营养素才能有效发挥作用,并建立包括构成呼吸系统的组织在内的组织。根据一些研究,诸如omega-3脂肪酸(亚麻酸、EPA,DHA)之类的健康脂肪有助于抑制肺部炎症,并为哮喘患者提供改善作用。
戒烟
如果您是吸烟者,那么可以改善呼吸系统健康的最重要的改变之一就是戒烟。香烟烟雾中的焦油和数千种化学物质往往会降低肺部的容量和效率。长期吸烟会导致不可逆的慢性呼吸问题。戒烟不仅有助于扭转急性健康问题,而且有助于改善呼吸系统健康。
运动
定期进行有氧运动可以帮助您改善呼吸健康。运动会增加肌肉对氧气的需求,导致大脑刺激呼吸系统,从而增加通风或呼吸。呼吸频率和呼吸深度的增加可扩展您的肺部,增强肺泡的弹性,排出肺部的旧空气,并增强隔膜。所有这些变化都具有持久作用,即使您停止运动后也可以帮助改善呼吸健康。
肥胖
肥胖是不良饮食和生活方式的结果,可能对呼吸系统造成不良影响。低胆固醇饮食对您的整体健康有益,而富含精制糖和反式脂肪的饮食会导致呼吸系统不良。
避免污染污染和城市烟雾可导致呼吸系统疾病,如哮喘,支气管炎和肺癌。如果您生活在大城市中,则戴上口罩会成为呼吸系统的防护屏障。
快餐
快餐可以增加您患呼吸系统疾病的机会。改用健康饮食以增强呼吸系统。
睡觉
几项研究表明,晚上多睡一小时的人患呼吸系统疾病和动脉阻塞钙化的风险较低,可导致心脏病。睡眠不足的人更容易出现呼吸系统问题。
酒精
喝酒会导致高血压,从而导致呼吸系统问题和心力衰竭。过量摄入酒精可能对您的整体健康有害。
避免过敏原
常见的过敏原,如尘螨,花粉,霉菌和动物皮屑,可能对呼吸系统有害。定期对所有表面进行真空吸尘。确保使用类似化学溶液定期清洁交流滤波器,窗帘和地毯。Tips for healthy respiratory system | Lung Diseases
https://www.onlymyhealth.com/health-slideshow/tips-for-healthy-respiratory-system-1390564565.html
Adv Pharm Bull. 2014 Dec; 4(Suppl 2): 555–561.
The Effect of Omega-3 Fatty Acids on ARDS: A Randomized Double-Blind Study
Masoud Parish, 1 Farnaz Valiyi, 1 Hadi Hamishehkar, 2 Sarvin Sanaie, 3 Mohammad Asghari Jafarabadi, 4 Samad EJ Golzari, 5 and Ata Mahmoodpoor 1 ,*
1 Faculty of Medicine, Tabriz University of Medical Sciences, Tabriz, Iran.
2 Applied Drug Research Center, Department of Clinical Pharmacy, Tabriz University of Medical Sciences, Tabriz, Iran.
3 Tuberculosis & Lung Research Center, Tabriz University of Medical Sciences, Tabriz, Iran.
4 Road Traffic Injury Research Center, Faculty of Health, Tabriz University of Medical Sciences, Tabriz, Iran.
5 Cardiovascular Research Center, Tabriz University of Medical Sciences, Tabriz, Iran.
Abstract
Purpose: The aim of this study was to evaluate the effect of an enteral nutrition diet, enriched with omega-3 fatty acids because of its anti-inflammatory effects on treatment of patients with mild to moderate ARDS.
Methods: This randomized clinical trial was performed in two ICUs of Tabriz University of Medical Sciences from Jun 2011 until Sep 2013 in north west of Iran. Fifty-eight patients with mild to moderate ARDS were enroled in this clinical trial. All patients received standard treatment for ARDS based on ARDS network trial. In intervention group, patients received 6 soft-gels of omega-3/day in addition to the standard treatment.
Results: Tidal volume, PEEP, pH, PaO2/FiO2 , SaO2, P platue and PaCO2 on the 7th and 14th days didn’t have significant difference between two groups. Indices of lung mechanics (Resistance, Compliance) had significant difference between the groups on the 14th day. Pao2 had significant difference between two groups on both 7th and 14th days. Trend of PaO2 changes during the study period in two groups were significant. We showed that adjusted mortality rate did not have significant difference between two groups.
Conclusion: It seems that adding omega-3 fatty acids to enteral diet of patients with ARDS has positive results in term of ventilator free days, oxygenation, lung mechanic indices; however, we need more multi center trials with large sample size and different doses of omega-3 fatty acids for their routine usage as an adjuant for ARDS treatment.
Keywords: ARDS, Inflammation, Omega-3 fatty acids, ICUThe Effect of Omega-3 Fatty Acids on ARDS: A Randomized Double-Blind Study
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4312405/
Lung Compliance and Elastance | Owlcation
https://owlcation.com/stem/Lung-Compliance-and-Elastance
The ability of the lungs to expand is expressed using a measure known as the lung compliance. Lung compliance is the volume change that could be achieved in the lungs per unit pressure change. Elastance, also known as the elastic resistance is the reciprocal of compliance, i.e. the pressure change that is required to elicit a unit volume change. This is a measure of the resistance of a system to expand.
Published: Jul 29, 2013What’s The Difference Between Oxygen Saturation And PaO2?
https://airwayjedi.com/2015/12/09/difference-oxygen-saturation-pao2
Pa02, put simply, is a measurement of the actual oxygen content in arterial blood. Partial pressure refers to the pressure exerted on the container walls by a specific gas in a mixture of other gases. When dealing with gases dissolved in liquids like oxygen in blood, partial pressure is the pressure that the dissolved gas would have if the blood were allowed to equilibrate with a volume of gas in a container. In other words, if a gas like oxygen is present in an air space like the lungs and also dissolved in a liquid like blood, and th…
Partial Pressure of Oxygen (PaO2) Test: Uses, Procedure ...
https://www.verywellhealth.com/partial-pressure-of-oyxgen-pa02-914920
Nov 19, 2019 · The partial pressure of oxygen, also known as PaO2, is a measurement of oxygen pressure in arterial blood. It reflects how well oxygen is able to move from the lungs to the blood, and it …
Omega 3 Fatty Acids May Reduce Bacterial Lung Infections Associated with COPD
Tuesday, March 15, 2016
Compounds derived from omega-3 fatty acids – like those found in salmon – might be the key to helping the body combat lung infections, according to researchers at the University of Rochester School of Medicine and Dentistry.
The omega-3 derivatives were effective at clearing a type of bacteria called Nontypeable Haemophilus influenzae (NTHi), which often plagues people with inflammatory diseases like chronic obstructive pulmonary disease (COPD).
COPD, which is most often caused by years of smoking, is characterized by inflammation and excessive mucus in the lungs that blocks airflow. Quitting can slow the progress of COPD, but it doesn’t halt the disease. Anti-inflammatory drugs are the most common treatment, however they suppress the immune system, which can put people with COPD at risk for secondary infections, most commonly NTHi bacterial infections.
Our biggest concern with patients who have COPD is bacterial infections, which often put their lives at risk. If we can figure out how to predict who is likely to get an infection, physicians could put them on a preventative medication.
“Our biggest concern with patients who have COPD is bacterial infections, which often put their lives at risk,” says Richard Phipps, Ph.D. professor of Environmental Medicine and director of the URSMD Lung Biology and Disease Program. “If we can figure out how to predict who is likely to get an infection, physicians could put them on a preventative medication.”
In his recent study, which was featured in the top ten percent of the March 15 issue of The Journal of Immunology, Phipps and lead author, Amanda Croasdell, a graduate student in the Toxicology program, tested the effectiveness of an inhalable omega-3 derivative to prevent NTHi lung infections in mice.
Omega-3 fatty acids, which are abundant in fish like sardines and salmon, are touted for their many health benefits. These superstars of the diet world are normally broken down to form molecules that help turn off inflammation after an infection or injury.
Richard Phipps, Ph.D.
“We never really knew why diets high in omega fatty acids seemed good, but now we know it’s because they provide the precursors for molecules that help shut down excessive inflammation.” says Phipps.
Doctors used to believe that shutting down inflammation only required removing whatever caused it, for example pulling a thorn from your finger or, in this case, getting rid of bacteria. While that might work some of the time, we now know that shutting down inflammation is an active process that requires a certain class of anti-inflammatory molecules.
Unlike other anti-inflammatory drugs, the specialized agent used in this study reduced inflammation in the lungs of mice without suppressing the ability to clear the bacteria. In fact, it could actually hasten the process of clearing bacteria. Phipps and his colleagues believe they are the first to show that this special compound can improve lung function in the face of live bacteria.
While these results are encouraging, further study is needed to understand how these compounds can be used in humans. A similar compound in the form of an eye drop solution was recently tested in a clinical trial for dry eye syndrome and was well tolerated.
If found to be effective in humans, the agent used in this study might have the potential to improve the lives of the millions of people around the world who suffer from COPD, and might also be used to treat ear infections, bronchitis, and pneumonia, which are also caused by NTHi.
###
The University of Rochester Medical Center is home to approximately 3,000 individuals who conduct research on everything from cancer and heart disease to Parkinson’s, pandemic influenza, and autism. Spread across many centers, institutes, and labs, our scientists have developed therapies that have improved human health locally, in the region, and across the globe. To learn more, visit http://www.urmc.rochester.edu/research.
欧米茄3脂肪酸可减少与COPD相关的细菌性肺部感染
2016年3月15日星期二
罗彻斯特大学医学中心(University of Rochester Medical Center)大约有3,000人,从事从癌症和心脏病到帕金森氏病,大流行性流感和自闭症的各种研究。我们的科学家分布在许多中心,研究所和实验室中,其开发的疗法改善了当地,该地区以及全球的人类健康。要了解更多信息,请访问http://www.urmc.rochester.edu/research。
欧米茄3脂肪酸可以减少与COPD相关的细菌性肺部感染-新闻室-罗切斯特大学医学中心
罗切斯特大学医学院(Rochester University of Medicine and Dentistry)的研究人员称,源自omega-3脂肪酸的化合物(如鲑鱼中的那些化合物)可能是帮助机体抵抗肺部感染的关键。
omega-3衍生物可有效清除一种称为非典型流感嗜血杆菌(NTHi)的细菌,这种细菌经常困扰患有慢性阻塞性肺疾病(COPD)等炎性疾病的人。
COPD(慢性阻塞性肺病)最常由多年吸烟引起,其特征是炎症和肺部粘液过多阻止气流。戒烟可以减慢COPD的进程,但并不能阻止这种疾病。抗炎药是最常见的治疗方法,但是它们会抑制免疫系统,使COPD患者处于继发感染(最常见的是NTHi细菌感染)的风险中。
对于患有COPD的患者,我们最大的担忧是细菌感染,这常常使他们的生命处于危险之中。如果我们能弄清楚如何预测谁可能感染,医生可以将其放在预防药物上。
“我们对患有COPD的患者最大的担忧是细菌感染,这常常使他们的生命处于危险之中,”理查德·菲普斯(Richard Phipps)博士说。他是环境医学教授,URSMD肺部生物与疾病计划主任。 “如果我们能弄清楚如何预测谁可能感染,医生可以将它们放在预防药物上。”
在他最近发表在3月15日出版的《免疫学杂志》中排名前十位的研究中,Phipps和主要作者Amanda Croasdell,毒理学项目的研究生,测试了可吸入omega-3衍生物的有效性预防小鼠的NTHi肺部感染。
鱼,沙丁鱼和鲑鱼中富含的Omega-3脂肪酸因其许多健康益处而受到吹捧。这些饮食界的超级巨星通常会分解形成有助于感染或受伤后关闭炎症的分子。
Phipps说:“我们从来不真正知道为什么富含欧米茄-3脂肪酸的饮食看起来不错,但是现在我们知道这是因为它们提供了有助于阻止过度炎症的分子的前体。”
医生过去认为,关闭炎症仅需消除引起炎症的原因,例如从手指上拔刺或在这种情况下去除细菌。尽管这有时可能会起作用,但我们现在知道,关闭炎症是一个活跃的过程,需要某种类型的抗炎分子。
与其他抗炎药不同,本研究中使用的特殊药物可减轻小鼠肺部的炎症,而不会抑制清除细菌的能力。实际上,它实际上可以加快清除细菌的过程。菲普斯和他的同事认为,他们是第一个证明这种特殊化合物在肺感染的情况下可以改善肺功能的药物。
尽管这些结果令人鼓舞,但需要进一步研究以了解这些化合物如何在人体中使用。滴眼液形式的类似化合物最近在临床试验中测试了干眼症,并且具有良好的耐受性。
如果发现对人类有效,则本研究中使用的药物可能会改善全球数百万患有COPD的人们的生活,也可能用于治疗耳部感染,支气管炎和肺炎,这也是由NTHi引起的。
Omega 3 Fatty Acids May Reduce Bacterial Lung Infections Associated with COPD - Newsroom - University of Rochester Medical Center
https://www.urmc.rochester.edu/news/story/4526/omega-3-fatty-acids-may-reduce-bacterial-lung-infections-associated-with-copd.aspx
Immunol Cell Biol. 2007 Apr-May;85(3):229-37. Epub 2007 Feb 20.
Pulmonary epithelial cells are a source of interferon-gamma in response to Mycobacterium tuberculosis infection.
Sharma M1, Sharma S, Roy S, Varma S, Bose M.
Author information
1
Department of Microbiology, VP Chest Institute, University of Delhi, Delhi, India.
Abstract
Recent report from our laboratory showed that A549 cells representing alveolar epithelial cells produce chemokine interleukin-8 and nitric oxide (NO) when challenged with Mycobacterium tuberculosis. Interferon-gamma (IFN-gamma) played a critical role in priming these cells to generate NO in vitro. In the present study, we report that M. tuberculosis-infected A549 cells are capable of elaborating IFN-gamma as shown by enzyme-linked immunosorbent assay and intracellular staining for IFN-gamma. Secretion profile indicated that M. tuberculosis-infected A549 released significantly high concentration of IFN-gamma at 48 and 72 h post-infection. Low level of IFN-gamma release was also seen to be induced by gamma-irradiated M. tuberculosis and subcellular components of M. tuberculosis. Cell surface receptor analysis showed that the M. tuberculosis-infected A549 cells expressed enhanced levels of IFN-gamma receptors. This observation suggests that the endogenously produced IFN-gamma in response to M. tuberculosis infection plays a role in intracellular regulation of innate immunity against intracellular pathogen such as M. tuberculosis. This observation is further strengthened by the fact that infected A549 cells expressed signal transducer and activator of transcription 1 (STAT1), an important mediator for IFN-gamma signaling pathway, leading to expression of inducible NO synthase and subsequent release of NO in sufficient concentration to be mycobactericidal. Our results show that production of IFN-gamma and enhanced expression of IFN-gamma receptors by infected A549 cells is a local phenomenon occurring as de novo intracellular activity, in response to M. tuberculosis infection. To the best of our knowledge, this is the first report to show that A549 cells interact actively with M. tuberculosis to produce IFN-gamma that might play an important role in innate immunity against tuberculosis.Pulmonary epithelial cells are a source of interferon-gamma in response to Mycobacterium tuberculosis infection. - PubMed - NCBI
https://www.ncbi.nlm.nih.gov/pubmed/17310225
Type I interferon response to extracellular bacteria in the airway epithelium
Affiliations
Department of Pediatrics, Columbia University, New York, NY, USA
The airway epithelium possesses many mechanisms to prevent bacterial infection. Not only does it provide a physical barrier, but it also acts as an extension of the immune system through the expression of innate immune receptors and corresponding effectors. One outcome of innate signaling by the epithelium is the production of type I interferons (IFNs), which have traditionally been associated with activation via viral and intracellular organisms.We discuss how three extracellular bacterial pathogens of the airway activate this intracellular signaling cascade through both surface components as well as via secretion systems, and the differing effects of type I IFN signaling on host defense of the respiratory tract.
1型干扰素对呼吸道上皮细胞外细菌的应答
呼吸道上皮具有多种防止细菌感染的机制。它不仅提供了一个物理屏障,而且通过表达先天免疫受体和相应的效应器来作为免疫系统的延伸。上皮细胞先天信号转导的一个结果是I型干扰素(IFNs)的产生,这种干扰素传统上与病毒和细胞内生物的激活有关。
我们讨论了呼吸道的三种细胞外致病菌如何通过表面成分和分泌系统激活这种细胞内信号级联,以及I型干扰素信号对呼吸道宿主防御的不同作用。Type I interferon response to extracellular bacteria in the airway epithelium: Trends in Immunology
https://www.cell.com/trends/immunology/fulltext/S1471-4906(11)00151-7
Alpha interferon is produced by white blood cells other than lymphocytes, beta interferon by fibroblasts, and gamma interferon by natural killer cells and cytotoxic T lymphocytes (killer T cells). All interferons inhibit viral replication by interfering with the transcription of viral nucleic acid.
Alpha interferon | biochemistry | Britannica
www.britannica.com/science/alpha-interferon
J Clin Immunol. Author manuscript; available in PMC 2011 May 1.
Influenza-Induced Production of Interferon-Alpha is Defective in Geriatric Individuals
David H. Canaday, Naa Ayele Amponsah, Leola Jones, Daniel J. Tisch, Thomas R. Hornick, and Lakshmi Ramachandracorresponding author
Author information Copyright and License information Disclaimer
David H. Canaday, Geriatric Research, Education and Clinical Center (GRECC), Cleveland VA Medical Center, Cleveland, OH 44106, USA; Division of Infectious Diseases, Department of Medicine, Case Western Reserve University, Cleveland, OH 44106, USA;
Abstract
Background
The majority of deaths (90%) attributed to influenza are in person’s age 65 or older. Little is known about whether defects in innate immune responses in geriatric individuals contribute to their susceptibility to influenza.
Objective
Our aim was to analyze interferon-alpha (IFN-alpha) production in peripheral blood mononuclear cells (PBMCs) isolated from young and geriatric adult donors, stimulated with influenza A or Toll-like receptor (TLR) ligands. IFN-alpha is a signature anti-viral cytokine that also shapes humoral and cell-mediated immune responses.
Results
Geriatric PBMCs produced significantly less IFN-alpha in response to live or inactivated influenza (a TLR7 ligand) but responded normally to CpG ODN (TLR9 ligand) and Guardiquimod (TLR7 ligand). All three ligands activate plasmacytoid dendritic cells (pDCs). While there was a modest decline in pDC frequency in older individuals, there was no defect in uptake of influenza by geriatric pDCs.
Discussion and Conclusion
Influenza-induced production of IFN-alpha was defective in geriatric PBMCs by a mechanism that was independent of reduced pDC frequency or viability, defects in uptake of influenza, inability to secrete IFN-alpha, or defects in TLR7 signaling.
Keywords: Aging, influenza, interferon-alpha, plasmacytoid dendritic cellsIntroduction
Over 32,000 deaths per year occur in a typical influenza season with over 90% of deaths in persons over 65 years of age [1]. With the current H1N1 global pandemic, the number of influenza cases is expected to dramatically increase resulting in greater numbers of deaths than usual even in individuals over age 65. They are less susceptible than younger individuals to 2009 H1N1 but continue to have greater risk of complications if they develop active influenza infection. Unfortunately, the seasonal influenza vaccine has poor efficacy in this older group. The rates of protection against hospitalization for influenza and pneumonia are estimated to be only 33% in individuals over age 65 [2]. A better understanding of the pathogenesis of influenza infection and disease in older persons is required to develop more effective vaccines or immunomodulatory strategies to reduce morbidity and mortality in this group.
There are likely multiple mechanisms for this increased morbidity and mortality with aging. While decline in T cell function has been extensively documented in the elderly [3–11], potential changes in immune function that impact innate anti-viral responses have been largely unexplored. Type I interferons (IFNs) were first identified for their antiviral properties against influenza [12, 13] and have been subsequently shown to induce the transcription of several genes that help degrade viral RNA and block viral replication [14–16]. In addition to their anti-viral properties, type I IFNs have multiple effects on human mononuclear populations including T cells and B cells, and reduced type I IFN production could result in decreased induction of cell-mediated immunity [17].
Plasmacytoid dendritic cells (pDCs) are the main producers of type I IFNs after influenza activation [18]. Influenza virus ssRNA induces expression of type I IFNs in pDCs via activation of Toll-like receptor 7 (TLR7) [19–21]. IFNs induce expression of MxA, a protein with anti-viral activity [22–24] that renders pDCs resistant to virus-induced apoptosis [25]. pDCs have been found in all of the relevant respiratory mucosal sites in humans including nasal mucosa, lung, and bronchalveolar lavage fluid [26–29]. In the influenza challenge mouse model, pDCs in the respiratory tract were found to make two-thirds of the IFN-alpha generated supporting the idea that pDCs in spite of their low numbers in respiratory mucosal sites produce the majority of IFN-alpha [30].
Several animal models suggest a clear role for IFN-alpha in protection against influenza. Ferrets and Guinea pigs have been found to be representative models of human disease as they are susceptible to human influenza strains, are able to transmit infection [31–33], and express the MxA gene [34]. Recent studies in ferrets and Guinea pigs show that exogenous IFN-alpha reduces viral shedding and morbidity to influenza A virus [35, 36]. The mouse model has shown variable results regarding the importance of IFN-alpha as many of the studies use BALB and B6 mice that lack the MxA gene [34].
In the current study, we have assessed response of peripheral blood mononuclear cells (PBMCs) isolated from control (<35 years) and geriatric (>65 years) individuals to live influenza. IFN-alpha production was significantly reduced in pDCs from geriatric individuals in response to influenza but not to other TLR ligands that also activate pDCs.
Results
PBMCs from Geriatric Individuals Produce Less IFN-Alpha in Response to Influenza than PBMCs from Younger Controls
Influenza Activates pDCs Via TLR7
Geriatric PBMCs Have Reduced Frequency of pDCs
Geriatric PBMCs are not Defective in Production of IFN-Alpha in Response to TLR9 Ligand CpG ODN or Other TLR7 Ligand Guardiquimod
Geriatric pDCs are Not Defective in Uptake of Influenza A
In conclusion, geriatric pDCs were defective in the production of IFN-alpha in response to influenza A by a mechanism that was independent of reduced pDC viability, defects in uptake of influenza, inability to secrete IFN-alpha, or defects in TLR7 signaling.Discussion
Susceptibility of older individuals to influenza has often been ascribed to defects in T cell function. The contribution of the innate immune response to this defect remains unclear. Innate immune responses to influenza consist importantly of vigorous production of type I IFNs. They are potent anti-viral cytokines that also regulate other aspects of innate and adaptive immunity. We observed a significant decrease in the levels of IFN-alpha in supernatants from geriatric PBMCs activated with influenza. Surprisingly, no defect in IFN-alpha production was observed in geriatric PBMCs activated with CpG ODN 2216 or Guardiquimod. While influenza activates pDCs via TLR7, CpG ODN 2216 and Guardiquimod activate pDCs via TLR9 and TLR7, respectively. Our observations clearly indicate no defect in TLR7 or TLR9 signaling or IFN-alpha production in the pDC population in the elderly. Consistent with our findings, Jing et al. recently reported that the frequency of IFN-alpha secreting pDCs was reduced in PBMCs from healthy elderly subjects activated with influenza but not CpG 2216 [47]. We also observed a decline in pDC frequency in the elderly similar to that reported by other groups [47– 49]. Less IFN-alpha secreting pDCs coupled with decline in pDC frequency in geriatric PBMCs may account for the decreased response to influenza in geriatric PBMCs. But geriatric pDCs would have to make more IFN-alpha on a per pDC basis in response to Guardiquimod and CpG ODN 2216 (Fig. 6) to account for their normal response to these ligands despite lower pDC frequency.
Defect in IFN-alpha production in geriatric pDCs was only observed with influenza but not with other ligands that also target intracellular TLRs in pDCs. Interaction of ligands with TLR7 and TLR9 occurs in acidic endocytic vesicles, is pH dependent and is abrogated by agents like chloroquine and bafilomycin A that increase endosomal pH [44–46]. Interaction of influenza viral ssRNA with TLR7 requires virus fusion and uncoating from endocytic vacuoles by a process that is pH dependent and occurs in late endosomes through a type I fusion process [53, 54]. Wang et al. demonstrated that increasing intraendosomal pH from approximately 4.5 to 5.2 with chloroquine significantly decreased IFN-alpha production in human pDCs in response to influenza virus but not to TLR7 ligand R848 that is an imidazoquinoline compound like Guardiquimod [55]. Increasing intraendosomal pH to 5.8 abrogated IFN-alpha production in response to both R848 and influenza. Therefore, subtle variations in late endosomal pH may impact IFN-alpha production in response to influenza but not other TLR7 ligands. We speculate that a slight increase in pH in late endosomes (and maybe all endosomal compartments) in the elderly may impact influenza virus fusion and uncoating and lead to inhibition of IFN-alpha production in response to influenza (which is very pH sensitive) but have little impact on IFN-alpha production to other TLR7 and TLR9 ligands (which may not be as pH sensitive).
The intracellular location of a TLR ligand may also determine the resulting biological response [56]. In human pDCs localization of CpG ODNs to transferrin-receptor-positive early endosomes led exclusively to IFN-alpha production while localization of CpG ODNs to LAMP-1 positive late endosomes promoted maturation of pDCs [56]. Similarly, in murine pDCs retention of Type “A” CpG ODN in endosomal compartments promoted IFN-alpha induction [57] while rapid transfer to lysosomal vesicles, as seen with conventional DCs, led to little IFN-alpha production. When Type “A” CpG ODN was manipulated for endosomal retention, robust production of IFN-alpha was observed [57]. In geriatric pDCs, viral ssRNA may be rapidly transferred to lysosomes, resulting in reduced IFN-alpha production.
Entry of influenza virus into cells has been studied extensively and involves binding of the virus to sialic acid-containing receptors on the cell surface followed by internalization by receptor-mediated endocytosis [58]. Since decreased uptake of influenza virus by geriatric pDCs may also lead to decreased IFN-alpha production, we compared uptake of FITC-labeled virus in geriatric and control pDC by flow cytometry. No defect in uptake of influenza by geriatric pDCs was observed. Although the experiment was designed with an incubation period to allow virus to bind cells, followed by a chase period to maximize uptake of virus, the possibility that virus remained on the cell surface cannot be ruled out.
In addition to their anti-viral properties, type I IFNs mediate both innate and adaptive immune responses. Therefore, reduced IFN-alpha levels in geriatric individuals after influenza stimulation could have a number of deleterious effects on both innate and adaptive immunity in older adults. Type I IFNs can induce the production of multiple cytokines, chemokines, and other molecules like IL-15 [59], CCXCL10, CCL4, CCL2, and IL-1RA [60]. Type I IFNs have been shown to play a modulatory role in differentiation of human T cells to Th1 development [61, 62] and in the development of CD8+ T central memory cells [63, 64]. Presence of IFN-alpha was shown to have mixed effects on proliferation in human memory CD4+ T cells depending on the antigen stimulation [65]. In murine systems, type I IFNs act directly on both CD4+ and CD8+ T cells to allow clonal expansion in response to viral infection [66, 67]. An in vitro study by Jego et al. [68] using human DCs and B cells showed that IFN induces activation and IL-6 secretion by DCs, which was required for differentiation of B cells into antibody-secreting cells. Isotype switching of B cells is also enhanced by type I IFNs [69]. In murine systems, following influenza virus infection, type I IFN receptor signals directly activated local B cells [70] and type I IFN directly modulated respiratory tract B cell responses [71]. Type I IFN has also been shown to enhance B cell receptor-dependent B cell responses [72]. A number of DC functions are enhanced by type I IFNs including MHC-I cross priming and DC maturation [73– 75]. Therefore, innate immune defects in IFN-alpha production by older individuals could lead to multiple defects in their adaptive immune responses.
Conclusions
Geriatric PBMCs made significantly less IFN-alpha in response to influenza than PBMCs from younger controls. This could have a deleterious effect on anti-influenza responses in geriatric individuals and contribute significantly to the susceptibility of older individuals to influenza.Influenza-Induced Production of Interferon-Alpha is Defective in Geriatric Individuals
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2875067/
Circulating, Interferon-Producing Plasmacytoid Dentritic Cells Decline During Human Ageing
Scandinavian Journal of Immunology, 2002
Increased frequency and severity of infections in the elderly have been taken as indicative of declining immune function. Dendritic cells (DCs), the most important antigen-presenting cells, play a central role in initiating and modulating immune responses. One type, DC2, arises from precursor plasmacytoid DCs (pDCs), a rare population of circulating blood cells, whose hallmark function is rapid and copious production of interferon-alpha (IFN-alpha) upon microbial challenge. We found significant decreases of the circulating pDCs during ageing in healthy adult humans, as defined both by flow cytometry and IFN-alpha generation. Mean pDC/mm3 in peripheral blood declined from 7.8 for the youngest age group (18-39 years) to 4.2 for the oldest (60-91 years; P = 0.017). IFN-alpha generation declined similarly, from 3537 to 1201 IU/ml, respectively (P = 0.006). There was also a slight decline over the age range in the amount of IFN generated per pDC (slope = -0.0087; P = 0.046). CD4+ T cells decreased by approximately 20% over the same age range (P = 0.001), while there was no change in the total lymphocyte or monocyte counts.
Circulating, Interferon-Producing Plasmacytoid Dendritic ...
https://www.researchgate.net/publication/11053876_Circulating_Interferon-Producing...In this regard, several studies using a single 57 or double 46 platform approach have demonstrated a significant and progressive reduction in the frequency of pDCs in peripheral blood, particularly...
Difference between INF Alpha, Beta and Gamma | easybiologyclass
https://www.easybiologyclass.com/difference-between-type-i-and-type-ii-interferons-inf-alphabeta-vs-inf-gamma/
Direct Effects of Type I Interferons on Cells of the Immune System
Sandra Hervas-Stubbs, Jose Luis Perez-Gracia, Ana Rouzaut, Miguel F. Sanmamed, Agnes Le Bon and Ignacio Melero
Abstract
Type I interferons (IFN-I) are well-known inducers of tumor cell apoptosis and antiangiogenesis via signaling through a common receptor interferon alpha receptor (IFNAR). IFNAR induces the Janus activated kinase–signal transducer and activation of transcription (JAK-STAT) pathway in most cells, along with other biochemical pathways that may differentially operate, depending on the responding cell subset, and jointly control a large collection of genes. IFNs-I were found to systemically activate natural killer (NK) cell activity. Recently, mouse experiments have shown that IFNs-I directly activate other cells of the immune system, such as antigen-presenting dendritic cells (DC) and CD4 and CD8 T cells. Signaling through the IFNAR in T cells is critical for the acquisition of effector functions. Cross-talk between IFNAR and the pathways turned on by other surface lymphocyte receptors has been described. Importantly, IFNs-I also increase antigen presentation of the tumor cells to be recognized by T lymphocytes. These IFN-driven immunostimulatory pathways offer opportunities to devise combinatorial immunotherapy strategies. Clin Cancer Res; 17(9); 2619–27. ©2011 AACR.Direct Effects of Type I Interferons on Cells of the Immune System | Clinical Cancer Research
https://clincancerres.aacrjournals.org/content/17/9/2619
Access denied | British Society for Immunology
https://www.immunology.org/public-information/bitesized-immunology/receptores-y-mol%C3%A9culas/interferons-type-i
PLoS Pathog. 2012 Jan; 8(1): e1002352.
Type 1 Interferons and Antiviral CD8 T-Cell Responses
Raymond M. Welsh, * Kapil Bahl, ¤ Heather D. Marshall, ¤ and Stina L. Urban
Glenn F. Rall, EditorDepartment of Pathology and Program in Immunology and Virology, University of Massachusetts Medical School, Worcester, Massachusetts, United States of America,
The Fox Chase Cancer Center, United States of America
Type 1 interferons (IFNs) were the first cytokines discovered and include IFNβ, >ten forms of IFNα, and several other related molecules that all bind to the same type 1 IFN receptor (IFN1R). Type 1 IFNs are commonly referred to as “viral” IFNs because they can be induced directly by virus infections, in contrast to “immune” IFN, or IFNγ, which is synthesized after receptor engagement of T cells and natural killer (NK) cells during immune responses. Type 1 IFNs get induced by viral nucleic acids and proteins acting on cellular signaling molecules such as Toll-like receptors and RNA helicases, which, in turn, release transcription factors into the nucleus. Mice lacking IFN1R appear normal in a pathogen-free environment but are extraordinarily susceptible to virus infections [1]. This susceptibility is partially due to IFN-regulated genes that suppress viral replication, but type 1 IFNs also have many immunoregulatory properties that could also affect host susceptibility to infection.
Indications of the immunoregulatory roles of type 1 IFN came in the 1970s with observations that IFN upregulated the expression of class 1 MHC antigens [2], enhanced histamine secretion by triggered Mast cells [3], and cytolytically activated NK cells [4]–[6]. Several studies showed that addition of IFN to mixed lymphocyte cultures could enhance or inhibit T-cell proliferation, depending on the dose [7]. IFN was then shown to elicit NK cell proliferation in vivo by a mechanism involving the induction of IL-15, a growth factor for NK cells [8], [9]; a similar phenomenon of IFN and IL-15 was later shown for the division of memory T cells [10]. In the past decade a substantial number of new insights have developed in regards to how IFN can directly or indirectly affect T-cell responses to viral infections. IFN can affect T-cell responses by acting on the antigen-presenting cells (APCs), by acting on the T cells, or by inducing other cytokines and chemokines that regulate T-cell responses. Of note is that the phenotype of the T cells and the timing of IFN exposure are of essence, as IFN can inhibit proliferation or induce apoptosis under some circumstances yet be dramatically stimulatory under other conditions. Depending on their activation status, T cells can change their expression levels of IFN1R and their expression of signaling molecules downstream from the IFN1R.Mechanisms of IFN Signaling and Gene Activation
All type 1 IFNs bind to a receptor of two chains, IFNαR1, which is constitutively bound to tyrosine kinase 2 (TYK2), and IFNαR2, which is constitutively bound to Janus kinase 1 (JAK1). Ligand binding induces dimerization of both receptor chains and the phosphorylation of TYK2, JAK1, and the intracellular tyrosine residues of each IFN1R chain [11]–[13]. The transphosphorylation of both chains by these kinases results in activation of signal transducers and activators of transcription (STATs) 1 and 2. These form complexes that are translocated into the nucleus and activate the transcription of a wide variety of genes regulated by IFN-stimulated response elements (ISRE) [14], [15]. Type 1 IFNs can limit CD8 T-cell expansion when acting through STAT1, but they can also activate other STATs and promote T-cell expansion when, for example, acting through STAT4 [16], [17]. Type 1 IFNs can also activate STAT 3 and 5, which can mediate antiapoptotic and promitogenic effects in T cells that escape the antimitotic effects of IFN by downregulating STAT1 after activation [13], [18].
Type 1 IFN plays a major role in the CD8 T-cell response to viral infection, and its effects are on both the APCs (Figure 1A and 1B) and on the T cells (Figure 1D). T cells that are exposed to their cognate peptide antigen presented in the context of MHC (pMHC) on APC-like dendritic cells (DCs) get costimulated through receptors such as CD28 and CD40 ligand and undergo a differentiation program associated with several cycles of division, the expression of the transcription factors t-bet and eomesodermin, followed by the acquisition of effector functions (Figure 1D). These effector functions include cytotoxicity associated with the synthesis of the cytolytic proteins like perforin and the ability to secrete antiviral cytokines such as IFNγ [19]–[22]. Type 1 IFN upregulates expression of both MHC and costimulatory molecules and in so doing can greatly affect the initiation of these T-cell responses (Figure 1A and 1D) [23]. Overall, there is dramatic upregulation of MHC even in nonprofessional APC throughout the host during the course of a viral infection [24].
Figure 1
Effect of type 1 IFN on T-cell activation, proliferation, and apoptosis.
This schematic shows the effects of type 1 IFN on antiviral CD8 T-cell responses. (A) A virus infects an APC and induces IFN, which upregulates MHC and costimulatory molecules. (B) Activated APCs migrate into the spleen and lymph nodes to present viral pMHC to T cells. (C) IFN promotes apoptosis of preexisting memory T cells, which are rapidly phagocytosed by CD8α+ DCs. (D) IFN directly promotes the proliferation of antigen (Ag)-specific CD8 T cells at the beginning of the response. (E) IFN indirectly enables late comer Ag-specific T cells to become immediate effectors, but directly inhibits proliferation. (F) After synchronized contraction, the host is left with a new population of memory T cells and a loss of preexisting memory cells.
Costimulation of CD8 T Cells by Type 1 IFN
Type 1 IFN can provide a major costimulatory effect in its own right by binding to the IFN1R on CD8 T cells and greatly augmenting their proliferation (Figure 1D) [17], [25], [26]. IFNγ, if present, can elicit a similar effect [27]; this was demonstrated in IFN1R bone marrow chimeric mice infected with lymphocytic choriomeningitis virus (LCMV), where the IFN1R+ CD8 cells greatly outgrew the IFN1R- CD8 T cells. Interestingly, this effect was much less profound with vaccinia virus, which is a poor type 1 IFN inducer. Vaccinia virus, however, is a good inducer of IL-12, and IL-12 seems to play a compensatory stimulatory role for T cells in that infection [28]. IFN 1 has potent growth-inhibitory and apoptotic properties, so one might be surprised about this direct augmentation of proliferation. However, as mentioned above, IFN 1–induced growth inhibition is in part mediated through STAT1, but antigen-activated CD8 T cells during LCMV infection downregulate STAT1 and get released from that block [29]. Mice lacking STAT1 experience a putative “nonspecific” proliferation of their CD8 T cells, so it is speculated that IFN 1 signaling through STAT1 may retard nonspecific proliferation and allow the antigen-specific T cells to develop. The action of IFN 1 through other STAT molecules can induce antiapoptotic effects and augment the proliferation of T cells.
Altered T-Cell Differentiation and Proliferation Caused by Out-of-Sequence Signaling
The timing of IFN exposure can greatly affect the T-cell differentiation pathway and the magnitude of the T-cell response. It is well established that exposure to IFNγ promotes the differentiation of CD4 T cells into IFNγ-secreting Th1 cells [30], [31], but here we are talking about a timing-dependent exposure of CD8 T cells to type 1 IFN. Exposure of naïve CD8 T cells to APC and IFN before exposure to cognate antigen upregulates the T-cell expression of eomesodermin and sensitizes T cells to enter an altered differentiation pathway on encounter with cognate antigen (Figure 1E) [32]. Instead of undergoing several divisions before exerting effector functions, these sensitized CD8 T cells retain a naïve antigenic phenotype but act like memory cells and develop effector-cell properties associated with cytokine production and cytolytic activity within 2–4 h. This is not due to a direct effect of IFN on the T cells, as it occurs even if T cells lack IFN1R. It is more likely due to IFN acting on the APCs, which need to express the restricting MHC molecule for the cognate peptide to sensitize the T cells to respond differently to the cognate peptide.
We propose that the enhanced expression of MHC- presenting self-peptide provides a low level stimulus to naïve T cells, enabling them to retain a naïve T-cell antigenic phenotype yet produce transcription factors that allow them to respond to cognate peptide like a memory T cell.
A common phenomenon occurring during the course of a viral infection is a transient immune deficiency whereby T cells respond poorly to T-cell mitogens in vitro and to challenge with nonviral antigens in vivo [33]; this is, in fact, why one should not get vaccinated during illness. Several phenomena could account for this deficiency, including growth of virus in T cells, impaired antigen presentation, competition for T-cell growth factors, and induction of activation-induced cell death in a Fas ligand-rich environment. However, we have recently shown that type 1 IFN itself may account for much of this immune suppression, if the T cells are exposed to the IFN before cognate antigen encounter (Figure 1E) [34]. Prior exposure to IFN before cognate antigen stimulus impairs the proliferation of T cells after the antigen stimulus, even in the presence of IFN acting as a costimulatory factor, and the inhibition of proliferation in this case requires IFN1R on the T cells. The molecular mechanism for this IFN-induced impairment of proliferation is unknown, but this is reminiscent of earlier work showing that NK cells become hyporesponsive to IFN-mediated activation after having received a prior IFN stimulus [35], [36].
Therefore, T cells that receive an IFN stimulus prior to cognate antigen exposure become sensitized to immediately become effector cells by an indirect IFN-dependent mechanism; but they undergo reduced proliferation by a direct IFN-dependent mechanism. Together these mechanisms may limit de novo T-cell responses in the midst of a viral infection and may aid in the synchronization of the contraction phase of the immune response, because T cells recruited late into the antiviral response would undergo reduced clonal expansion.
IFN-Induced Apoptosis and Attrition of Memory T Cells
IFN-inducing viral infections have a deleterious effect on memory CD8 and CD4 T cells specific to other antigens. We show here that memory-phenotype CD8 T cells express moderately higher levels of IFN1R than do naïve T cells (Figure 2), and it is not unusual for 50%–80% of the memory CD8 T cells to undergo an IFN-induced apoptosis early during infection (Figure 1C) [37]–[40]. Some naïve cells also die in the earlier stages of infection, but to a much lower extent. This apoptosis is associated with elevated caspases, annexin V-staining, and DNA fragmentation and is at least partially dependent on Bim, known to be a proapoptotic molecule induced by type 1 IFN [39], [41]. Of note is that type 1 IFN inducers drive a substantial increase in the number of the highly phagocytic CD8α+, CD11c+ DC population into the spleen of mice (Figure 1B and 1C) [39]. These DC assimilate apoptotic cells and become reactive with Annexin V in the process, making it difficult to quantify apoptotic T cells directly ex vivo and easy to confuse CD8+ T cells with CD8+ DC. The IFN-induced apoptosis of memory T cells can occur in the presence of cognate antigen [38], leading one to question why such a mechanism should exist, as one might want to rapidly recruit antigen-specific memory cells into an immune response. One possibility is that this loss in memory cells is well tolerated because of their initial high frequencies and that it creates room for new T-cell responses to vigorously develop. It has been known for decades that partial depletion of lymphocyte populations can augment new T-cell responses [42], [43]. Further, should these memory T cells cross-react with another pathogen, a reduction in their number may prevent them from overzealously dominating the T-cell response to the cross-reactive epitope [38]. This IFN-induced loss in memory T cells at the beginning of infections would allow for a more diverse and presumably more effective T-cell response to that pathogen. Memory T cells may often be present in clonal excess such that the host can reduce their numbers without deleterious effects. However, a series of infections with heterologous pathogens has been shown to reduce memory T-cell numbers to levels that compromise the host's resistance to infections [44], [45].
Figure 2
Higher type 1 IFN R (IFNAR1) expression on CD44 high memory phenotype CD8 T cells.
Isolated spleen leukocytes from wild-type (WT) or IFNR knockout (KO) mice were stained with fluorescently labeled monoclonal antibodies (mAb) specific for CD8 (53-6.7; BD Pharmingen), CD44 (IM7; BD Pharmingen), and IFNAR-1 (MAR1-5A3; BioLegend). Stained samples were acquired using a BD Biosciences LSR II flow cytometer with FACS Diva software and analyzed with FlowJo software. The mean fluorescence intensity (MFI) for IFNAR1 is shown for CD44 low and CD44 high CD8 T cells, n = 3/group. **, p<0.005.
Conclusion: Sequence of Type 1 IFN–Induced Events during a Viral Infection
We now can envisage the series of type 1 IFN–induced events that control CD8 T-cell responses to viral infections (Figure 1). A virus will infect a host and possibly a DC and induce IFN that upregulates MHC and costimulatory molecules, and then the activated DC migrates into the spleen and lymph nodes (Figure 1A and 1B). IFN induces the apoptosis of many of the memory cells and some of the naïve cells, making room in the immune system to drive a strong T-cell response (Figure 1C). The antigen-specific T cells downregulate the antiproliferative STAT1, allowing IFN signals to go through other STAT molecules that inhibit apoptosis and promote proliferation (Figure 1D). Type 1 IFN acts as a strong costimulatory factor driving T-cell expansion. Late comer T cells in the immune response will be indirectly sensitized by IFN to immediately become effector cells but at the expense of proliferation, which is suppressed by direct IFN signaling (Figure 1E). After the virus is cleared, the T-cell response synchronously contracts, leaving the host with a pool of new memory cells and a loss of previously existing ones (Figure 1F).Type 1 Interferons and Antiviral CD8 T-Cell Responses
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3252364/
The major histocompatibility (MHC) class I antigen presentation pathway plays an important role in alerting the immune system to virally infected cells. MHC class I molecules are expressed on the cell surface of all nucleated cells and present peptide fragments derived from intracellular proteins.
The MHC class I antigen presentation pathway: strategies ...
www.ncbi.nlm.nih.gov/pmc/articles/PMC1783040/
Alveolar Type II Epithelial Cells Contribute to the Anti ...
https://mbio.asm.org/content/7/3/e00276-16
Jul 06, 2016 · Influenza A virus (IAV) periodically causes substantial morbidity and mortality in the human population. In the lower lung, the primary targets for IAV replication are type II alveolar epithelial cells (AECII), which are increasingly recognized for their immunological potential.
Cited by: 22
Publish Year: 2016
Author: S. Stegemann-Koniszewski, Andreas Jeron, Marcus Gereke, Robert Geffers, Andrea Kröger, Matthias Gunz...
Apoptosis and Pathogenesis of Avian Influenza A (H5N1 ...
https://wwwnc.cdc.gov/eid/article/13/5/06-0572
Infection of epithelial cells and lymphocytes has been shown to induce apoptosis in vitro (4–8). Several modes of apoptosis induction and responsible viral genes have been proposed (8–13). Infection with virulent influenza (H5N1) virus was also shown to induce lymphopenia and lymphocyte apoptosis in …
Alveolar epithelial cell injury with Epstein-Barr virus ...
https://www.physiology.org/doi/10.1152/ajplung.00376.2007
Idiopathic pulmonary fibrosis (IPF) is a refractory and lethal interstitial lung disease characterized by alveolar epithelial cells apoptosis, fibroblast proliferation, and ECM protein deposition. Epstein-Barr virus (EBV) has previously been localized to alveolar epithelial cells of IPF …
Cited by: 54
Publish Year: 2008
Author: Andrea P. Malizia, Dominic T. Keating, Sinead M. Smith, Dermot Walls, Peter P. Doran, Jim J. Egan
Respiratory Epithelial Cells as Master Communicators ...
https://link.springer.com/article/10.1007/s40588-019-0111-8
Feb 13, 2019 · Infected epithelial cells release exosomes that specifically regulate responses of monocytes and neighboring epithelial cells without promoting spread of virus. In contrast, rhinovirus-infected cells induce monocytes to upregulate expression of the viral receptor, promoting spread of the virus to alternate cell types.
Author: Tanya A. Miura
Location: Moscow
Publish Year: 2019
Influenza virus damages the alveolar barrier by disrupting epithelial cell tight junctions
Kirsty R. Short1,2, Jennifer Kasper3
, Stijn van der Aa1
, Arno C. Andeweg1
,
Fatiha Zaaraoui-Boutahar1
, Marco Goeijenbier1
, Mathilde Richard1
,
Susanne Herold4
, Christin Becker4
, Dana P. Scott5
, Ronald W.A.L. Limpens6
,
Abraham J. Koster6
, Montserrat Bárcena6
, Ron A.M. Fouchier1
,
Charles James Kirkpatrick3 and Thijs Kuiken1
Affiliations: 1
Dept of Viroscience, Erasmus Medical Center, Rotterdam, The Netherlands. 2
School of
Biomedical Sciences, University of Queensland, Brisbane, Australia. 3
Institute of Pathology, University Medical
Center, Johannes Gutenberg University, Mainz, Germany. 4
University of Giessen and Marburg Lung Center
(UGMLC), Justus-Liebig-University of Giessen, Member of the German Center for Lung Research (DZL),
Giessen, Germany. 5
Rocky Mountain Veterinary Branch, Division of Intramural Research, National Institute of
Allergy and Infectious Diseases, National Institutes of Health, Hamilton, MT, USA. 6
Dept of Molecular Cell
Biology, Section Electron Microscopy, Leiden University Medical Centre, Leiden, The Netherlands.
Correspondence: Thijs Kuiken, Dept of Viroscience, Erasmus Medical Center, Dr Molewaterplein 50, 3015GE
Rotterdam, The Netherlands. E-mail: t.kuiken@erasmusmc.nl
ABSTRACTA major cause of respiratory failure during influenza A virus (IAV) infection is damage to
the epithelial–endothelial barrier of the pulmonary alveolus. Damage to this barrier results in flooding of
the alveolar lumen with proteinaceous oedema fluid, erythrocytes and inflammatory cells. To date, the
exact roles of pulmonary epithelial and endothelial cells in this process remain unclear.
Here, we used an in vitro co-culture model to understand how IAV damages the pulmonary epithelial–
endothelial barrier. Human epithelial cells were seeded on the upper half of a transwell membrane while
human endothelial cells were seeded on the lower half. These cells were then grown in co-culture and IAV
was added to the upper chamber.
We showed that the addition of IAV (H1N1 and H5N1 subtypes) resulted in significant barrier damage.
Interestingly, we found that, while endothelial cells mounted a pro-inflammatory/pro-coagulant response
to a viral infection in the adjacent epithelial cells, damage to the alveolar epithelial–endothelial barrier
occurred independently of endothelial cells. Rather, barrier damage was associated with disruption of tight
junctions amongst epithelial cells, and specifically with loss of tight junction protein claudin-4.
Taken together, these data suggest that maintaining epithelial cell integrity is key in reducing pulmonary
oedema during IAV infection.
Influenza virus damages the alveolar barrier by disrupting epithelial cell tight junctions
https://erj.ersjournals.com/content/erj/47/3/954.full.pdf
Reactive oxygen and nitrogen species during viral infections
July 2014 Free Radical Research 48(10):1-29
University of Milan | UNIMI · Department of Pathophysiology and Transplantation
Biotechnologist, PhDAbstract
Oxygen and nitrogen radicals are frequently produced during viral infections.
These radicals are not only a physiological mechanism for pathogen clearance but also result in many pathological consequences. Low concentrations of radicals can promote viral replication; however high concentrations of radicals can also inhibit viral replication and are detrimental to the cell due to their mitogenic activity.
We reviewed the detailed mechanisms behind oxygen and nitrogen radical production and focused on how viruses induce radical production. In addition, we examined the effects of oxygen and nitrogen radicals on both the virus and host. We also reviewed enzymatic and chemical detoxification mechanisms and recent advances in therapeutic antioxidant applications. Many molecules that modulate the redox balance have yielded promising results in cell and animal models of infection. This encourages their use in clinical practice either alone or with existing therapies.
However, since the redox balance also plays an important role in host defence against pathogens, carefully designed clinical trials are needed to assess the therapeutic benefits and secondary effects of these molecules and whether these effects differ between different types of viral infections.
Reactive oxygen and nitrogen species during viral infections | Request PDF
https://www.researchgate.net/publication/264090810_Reactive_oxygen_and_nitrogen_species_during_viral_infections
The Influenza Virus H5N1 Infection Can Induce ROS ...
https://www.mdpi.com/1999-4915/8/1/13
The Influenza Virus H5N1 Infection Can Induce ROS Production for Viral Replication and Host Cell Death in A549 Cells Modulated by Human Cu/Zn Superoxide Dismutase (SOD1) Overexpression by Xian Lin 1,† , Ruifang Wang 1,† , Wei Zou 1 , Xin Sun 1 , Xiaokun Liu 1 , Lianzhong Zhao 1 …
Cited by: 24
Publish Year: 2016
Author: Xian Lin, Ruifang Wang, Wei Zou, Xin Sun, Xiaokun Liu
The Influenza Virus H5N1 Infection Can Induce ROS Production for Viral Replication and Host Cell Death in A549 Cells Modulated by Human Cu/Zn Superoxide Dismutase (SOD1) Overexpression
by Xian Lin 1,†, Ruifang Wang 1,†, Wei Zou 1, Xin Sun 1, Xiaokun Liu 1, Lianzhong Zhao 1, Shengyu Wang 1 and Meilin Jin 1,2,*
1
State Key Laboratory of Agricultural Microbiology, Huazhong Agricultural University, Wuhan 430070, China
2
Laboratory of Animal Virology, College of Veterinary Medicine, Huazhong Agricultural University, Wuhan 430070, China
*
Author to whom correspondence should be addressed.
†
These authors contributed equally to this work.
Academic Editor: Andrew Mehle
Viruses 2016, 8(1), 13; https://doi.org/10.3390/v8010013
Abstract
Highly pathogenic H5N1 infections are often accompanied by excessive pro-inflammatory response, high viral titer, and apoptosis; as such, the efficient control of these infections poses a great challenge. The pathogenesis of influenza virus infection is also related to oxidative stress. However, the role of endogenic genes with antioxidant effect in the control of influenza viruses, especially H5N1 viruses, should be further investigated.In this study, the H5N1 infection in lung epithelial cells decreased Cu/Zn superoxide dismutase (SOD1) expression at mRNA and protein levels. Forced SOD1 expression significantly inhibited the H5N1-induced increase in reactive oxygen species, decreased pro-inflammatory response, prevented p65 and p38 phosphorylation, and impeded viral ribonucleoprotein nuclear export and viral replication. The SOD1 overexpression also rescued H5N1-induced cellular apoptosis and alleviated H5N1-caused mitochondrial dysfunction. Therefore, this study described the role of SOD1 in the replication of H5N1 influenza virus and emphasized the relevance of this enzyme in the control of H5N1 replication in epithelial cells. Pharmacological modulation or targeting SOD1 may open a new way to fight H5N1 influenza virus.
Viruses | Free Full-Text | The Influenza Virus H5N1 Infection Can Induce ROS Production for Viral Replication and Host Cell Death in A549 Cells Modulated by Human Cu/Zn Superoxide Dismutase (SOD1) Overexpression
https://www.mdpi.com/1999-4915/8/1/13
Br J Pharmacol. 1999 Feb; 126(3): 730–734.
Effects of vitamin C and of a cell permeable superoxide dismutase mimetic on acute lipoprotein induced endothelial dysfunction in rabbit aortic rings
L Fontana,1 K L McNeill,1 J M Ritter,1 and P J Chowienczyk1,*Department of Clinical Pharmacology, Guy's, King's and St Thomas' School of Biomedical Sciences, St Thomas' Hospital, Lambeth Palace Road, London SE1 7EH, England
Abstract
Low density lipoprotein (LDL) inhibits endothelium-dependent relaxation. The mechanism is uncertain, but increased production of superoxide anion O2− with inactivation of endothelium-derived NO and formation of toxic free radical species have been implicated. We investigated effects of the cell permeable superoxide dismutase mimetic manganese (III) tetrakis (1-methyl-4-pyridyl) porphyrin (MnTMPyP), the free radical scavenger vitamin C and arginine (which may reduce O2− formation) on acute LDL-induced endothelial dysfunction in rabbit aortic rings, using LDL prepared by ultracentrifugation of plasma from healthy men and aortic rings from New Zealand white rabbits.
LDL (150 μg protein ml−1 for 20 min) markedly inhibited relaxation of aortic rings (in Krebs' solution at 37°C and pre-constricted to 80% maximum tension with noradrenaline) to acetylcholine 82±10% (mean percentage difference between sum of relaxations after each concentration of acetylcholine in the presence and absence of LDL, ±s.e.mean, n=26, P<0.001) but not to the endothelium-independent agonist nitroprusside.
MnTMPyP (10 μm) reduced inhibitory effects of LDL from 124±27 to 56±17% (n=6, P<0.05).
Vitamin C (1 mm) reduced inhibitory effects of LDL from 59±8 to 22±5% (n=6, P<0.05).
Inhibitory effects of LDL were similar in the absence or presence of arginine (84±12 vs 79±16%, n=14, P=0.55). Effects of l-arginine (10 mm) did not differ significantly from those of d-arginine (10 mm).
Acute (20 min) exposure of aortic rings to LDL impairs endothelium-dependent relaxation which can be partially restored by MnTMPyP and vitamin C. This is consistent with LDL causing increased O2− generation.
Keywords: Antioxidants, arginine, endothelium, low density lipoprotein, nitric oxide, oxidative stress, superoxide anion, superoxide dismutase, vitamin CExperimental Protocols
...
Similar experiments were performed using vitamin C (1 mm), l-arginine (10 mm) and d-arginine (10 mm). A similar protocol was also used to examine effects of LDL on relaxation to nitroprusside as a non endothelium-dependent control. Doses of MnTMPyP and vitamin C were chosen as the maximum dose which, in pilot studies, had no inhibitory effects on relaxation to acetylcholine.
Effects of vitamin C and of a cell permeable superoxide dismutase mimetic on acute lipoprotein induced endothelial dysfunction in rabbit aortic rings
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1565839/
World J Gastroenterol. 2003 Nov 15; 9(11): 2565–2569.
Therapeutic efficacy of high-dose vitamin C on acute pancreatitis and its potential mechanisms
Wei-Dong Du, Zu-Rong Yuan, Jian Sun, Jian-Xiong Tang, Ai-Qun Cheng, Da-Ming Shen, Chun-Jin Huang, Xiao-Hua Song, Xiao-Feng Yu, and Song-Bai Zheng
上海华东医院外科 消化内科
Abstract
AIM: To observe the therapeutic efficacy of high-dose Vitamin C (Vit. C) on acute pancreatitis (AP), and to explore its potential mechanisms.
METHODS: Eghty-four AP patients were divided into treatment group and control group, 40 healthy subjects were taken as a normal group. In the treatment group, Vit. C (10 g/d) was given intravenously for 5 d, whereas in the control group, Vit. C (1 g/d) was given intravenously for 5 d. Symptoms, physical signs, duration of hospitalization, complications and mortality rate were monitored. Meanwhile, serum amylase, urine amylase and leukocyte counts were also determined. The concentration of plasma vitamin C (P-VC), plasma lipid peroxide (P-LPO), plasma vitamin E (P-VE), plasma β-carotene (P-β-CAR), whole blood glutathione (WB-GSH) and the activity of erythrocyte surperoxide dimutase (E-SOD) and erythrocyte catalase (E-CAT) as well as T lymphocyte phenotype were measured by spectrophotometry in the normal group and before and after treatment with Vit. C in the treatment and the control group.
RESULTS: Compared with the normal group, the average values of P-VC, P-VE, P-β-CAR, WB-GSH and the activity of E-SOD and E-CAT in AP patients were significantly decreased and the average value of P-LPO was significantly increased, especially in severe acute pancreatitis (SAP) patients (P < 0.05. P-VC, P = 0.045; P-VE, P = 0.038; P = 0.041; P-β-CAR, P = 0.046; WB-GSH, P = 0.039; E-SOD, P = 0.019; E-CAT, P = 0.020; P-LPO, P = 0.038). Compared with the normal group, CD3 and CD4 positive cells in AP patients were significantly decreased. The ratio of CD4/CD8 and CD4 positive cells were decreased, especially in SAP patients (P < 0.05. CD4/CD8, P = 0.041; CD4, P = 0.019). Fever and vomiting disappeared, and leukocyte counts and amylase in urine and blood become normal quicker in the treatment group than in the control group. Moreover, patients in treatment group also had a higher cure rate, a lower complication rate and a shorter in-ward days compared with those in he control group. After treatment, the average value of P-VC was significantly higher and the values of SIL-2R, TNF-α, IL-6 and IL-8 were significantly lower in the treatment group than in the control group (P < 0.05 P-VC, P = 0.045; SIL-2R, P = 0.012; TNF-α, P = 0.030; IL-6, P = 0.015; and IL-8, P = 0.043). In addition, the ratio of CD4/CD8 and CD4 positive cells in the patients of treatment group were significantly higher than that of the control group after treatment (P < 0.05. CD4/CD8, P = 0.039; CD4, P = 0.024).
CONCLUSION: High-dose vitamin C has therapeutic efficacy on acute pancreatitis. The potential mechanisms include promotion of anti-oxidizing ability of AP patients, blocking of lipid peroxidation in the plasma and improvement of cellular immune function.
Ultrastructure of the lung in a murine model of malaria-associated acute lung injury/acute respiratory distress syndrome.
Aitken EH, Negri EM, Barboza R, Lima MR, Álvarez JM, Marinho CR, Caldini EG, Epiphanio S - Malaria journal (2014)
Background: The mechanisms through which infection with Plasmodium spp. result in lung disease are largely unknown. Recently a number of mouse models have been developed to research malaria-associated lung injury but no detailed ultrastructure studies of the disease in its terminal stages in a murine model have yet been published. The goal was to perform an ultrastructural analysis of the lungs of mice that died with malaria-associated acute lung injury/acute respiratory distress syndrome to better determine the relevancy of the murine models and investigate the mechanism of disease. Methods: DBA/2 mice were infected with Plasmodium berghei strain ANKA. Mice had their lungs removed immediately after death, processed using standard methods and viewed by transmission electron microscopy (TEM). Results: Infected red blood cell:endothelium contact, swollen endothelium with distended cytoplasmic extensions and thickening of endothelium basement membrane were observed. Septa were thick and filled with congested capillaries and leukocytes and the alveolar spaces contained blood cells, oedema and cell debris. Conclusion: Results show that the lung ultrastructure of P. berghei ANKA-infected mice has similar features to what has been described in post-mortem TEM studies of lungs from individuals infected with Plasmodium falciparum. These data support the use of murine models to study malaria-associated acute lung injury.
Diagram showing main findings of the paper. Using scann | Open-i
https://openi.nlm.nih.gov/detailedresult?img=PMC4062769_1475-2875-13-230-10&req=4
Education | July 2014
Mechanical Ventilation–associated Lung Fibrosis in Acute Respiratory Distress Syndrome: A Significant Contributor to Poor Outcome
Nuria E. Cabrera-Benitez, Ph.D.; John G. Laffey, M.D.; Matteo Parotto, M.D., Ph.D.; Peter M. Spieth, M.D., Ph.D.; Jesús Villar, M.D., Ph.D.; et al
Department of Anesthesia, University of Toronto, Toronto, Ontario, Canada.
Mechanical Ventilation–associated Lung Fibrosis in Acute Respiratory Distress Syndrome: A Significant Contributor to Poor Outcome Nuria E. Cabrera-Benitez, Ph.D.; John G. Laffey, M.D.; Matteo Parotto, M.D., Ph.D.; Peter M. Spieth, M.D., Ph.D.; Jesús Villar, M.D., Ph.D.; et al Abstract One of the most challenging problems in critical care medicine is the management of patients with the acute respiratory distress syndrome. Increasing evidence from experimental and clinical studies suggests that mechanical ventilation, which is necessary for life support in patients with acute respiratory distress syndrome, can cause lung fibrosis, which may significantly contribute to morbidity and mortality. The role of mechanical stress as an inciting factor for lung fibrosis versus its role in lung homeostasis and the restoration of normal pulmonary parenchymal architecture is poorly understood.
In this review, the authors explore recent advances in the field of pulmonary fibrosis in the context of acute respiratory distress syndrome, concentrating on its relevance to the practice of mechanical ventilation, as commonly applied by anesthetists and intensivists. The authors focus the discussion on the thesis that mechanical ventilation—or more specifically, that ventilator-induced lung injury—may be a major contributor to lung fibrosis. The authors critically appraise possible mechanisms underlying the mechanical stress–induced lung fibrosis and highlight potential therapeutic strategies to mitigate this fibrosis.
Mechanical ventilation may be a major contributor to pulmonary fibrosis in patients with the acute respiratory distress syndrome.
THE acute respiratory distress syndrome (ARDS) is a major cause of mortality.1 ARDS is characterized by its acute onset, bilateral pulmonary infiltrates, severe hypoxemia, and pulmonary edema of noncardiac origin.2–5 Pronounced morphological changes occur in the lung parenchyma and are associated with impaired lung function, which is partly reversible.5
Mechanical ventilation is the most important supportive therapy for patients with ARDS, but it can induce or aggravate lung injury—an entity referred to as ventilator-induced lung injury (VILI).6,7
ARDS is also characterized pathologically by an early exudative, inflammatory phase, followed in many patients by a fibrotic phase. The inflammatory phase is the focus of more studies—a PubMed search for ARDS AND inflammationyielded 561 articles, whereas a search for ARDS AND fibrosis yielded 260 articles.
In this review, we explore recent advances in the field of pulmonary fibrosis in the context of ARDS, concentrating on its relevance to the practice of mechanical ventilation, as commonly applied by anesthetists and intensivists. We focus our discussion on the thesis that mechanical ventilation—or more specifically, that VILI—may be a major contributor to lung fibrosis. We critically appraise possible mechanisms underlying the mechanical stress–induced lung fibrosis and highlight potential therapeutic strategies to mitigate this fibrosis.
Clinical Evidence of Lung Fibrosis in ARDS
Many patients with ARDS survive the acute phase, but subsequently go on to die, often with evidence of significant pulmonary fibrosis.8 Severe fibrosis was demonstrated to be a frequent complication in ARDS as early as the 1990s.9 Lung histologic studies of patients with late ARDS suggested ongoing inflammatory injury together with progressive fibrosis.10–12 Areas of exudation are found adjacent to advanced fibrosis, and epithelial and endothelial injury is pronounced in the late phase of ARDS10–12 (fig. 1).
Fig. 2. Mechanical stretch impairs alveolar epithelial integrity. The alveolar epithelial tight junction is consists of several constituents of connected proteins. Occludin is a transmembrane protein known to be associated with F-actin, either directly or indirectly modulating the tight junction structure. Mechanical stretch of alveolar epithelial cells can result in loss of tight junction structure and cell–cell attachment associated with decrease in the expression or increase in degradation of occludin and actin perturbations. The actin cytoskeleton remodeling plays an important role in fibrosis formation in the lung. ATI = alveolar type I; ATII = alveolar type II.
Fig. 3. Mechanical stretch causes inflammatory responses associated with release of mediators that can worsen lung injury leading to “biotrauma.” Mechanical stretch of alveoli results in increased expression of small fragment hyaluronan (sHA) and activation of cytoplasmic proline-rich tyrosine kinase-2 (PyK2); polymorphonuclear leukocyte (PMN) infiltration that release soluble mediators such as cytokines and platelet-derived growth factor (PDGF); increased production of extracellular matrix (ECM) proteins including transforming growth factor-β1 (TGF-β1), collagen, elastin, fibronectin laminin, lumican, proteoglycan, and glycosaminoglycans. During the exudative phase of acute respiratory distress syndrome, the influx of T regulatory cells (Treg) may play a critical role in the crosstalk between innate and adaptive immune systems that normally would modulate the transition from injury to repair in resolving lung injury. ATI = alveolar type I; ATII = alveolar type II.
Fig. 5. Potential mechanisms of mesenchymal stromal cells (MSCs) in the lung repair process in acute respiratory distress syndrome. MSCs exert a number of properties to enhance repair and restoration of physiologic function after ventilator-induced lung injury. The effects seem to be paracrine mediated and dependent in part on keratinocyte growth factor produced by the stromal cells. The bone marrow–derived MSC could transfer their mitochondria into lung epithelial cells resulting in increased alveolar adenosine triphosphate concentrations and enhanced cellular bioenergetics and improved lung function. The MSC may also be able to differentiate into alveolar type I (ATI) and type II (ATII) epithelial cells.
Conclusions
Recent evidence demonstrates that mechanical ventilation, particularly where significant overstretch occurs, may drive the pathogenesis of fibrosis in patients with ARDS. The application of mechanical ventilation in animal models of acute lung injury or the application of mechanical stress in vitro in lung epithelial cells can induce the development of lung fibrosis through fibroproliferation and EMT. Future studies are required to improve our understanding of these mechanisms so that we can develop novel approaches—pharmacologic or other—to prevent or treat the pulmonary fibrosis associated with mechanical ventilation in patients with ARDS.
Mechanical Ventilation–associated Lung Fibrosis in Acute Respiratory Distress Syndrome:A Significant Contributor to Poor Outcome | Anesthesiology | ASA Publications
https://anesthesiology.pubs.asahq.org/article.aspx?articleid=1917689
Influenza-induced monocyte-derived alveolar macrophages ...
https://www.nature.com/articles/s41590-019-0568-x
Jan 13, 2020 · Fig. 7: Monocyte-derived AMs persist for 2 months after infection with influenza, but do not produce increased IL-6 and afford bacterial protection. Data …
Author: Helena Aegerter, Justina Kulikauskaite, Stefania Crotta, Harshil Patel, Gavin Kelly, Edith M. Hessel...
Publish Year: 2020
Monocyte-derived IL-1 and IL-6 are differentially required ...
https://www.ncbi.nlm.nih.gov/pubmed/29808007
In humanized mice with high leukemia burden, CAR T cell-mediated clearance of cancer triggered high fever and elevated IL-6 levels, which are hallmarks of CRS. Human monocytes were the major source of IL-1 and IL-6 during CRS. Accordingly, the syndrome was prevented by monocyte depletion or by blocking IL-6 receptor with tocilizumab.
Cited by: 192
Publish Year: 2018
Author: Margherita Norelli, Barbara Camisa, Giulia Barbie
The innate immune architecture of lung tumors and its implication in disease progression - Milette - 2019 - The Journal of Pathology - Wiley Online Library
https://onlinelibrary.wiley.com/doi/10.1002/path.5241
Nat Rev Immunol. Author manuscript; available in PMC 2016 Mar 8.
The role of airway epithelial cells and innate immune cells in chronic respiratory disease
Michael J. Holtzman,1,2 Derek E. Byers,1 Jennifer Alexander-Brett,1 and Xinyu Wang1
Author information Copyright and License information Disclaimer
1Pulmonary and Critical Care Medicine, Department of Medicine, Washington University School of Medicine, Saint Louis, Missouri 63110, USA.
2Department of Cell Biology, Washington University School of Medicine, Saint Louis, Missouri 63110, USA.
Abstract
An abnormal immune response to environmental agents is generally thought to be responsible for causing chronic respiratory diseases, such as asthma and chronic obstructive pulmonary disease (COPD). Based on studies of experimental models and human subjects, there is increasing evidence that the response of the innate immune system is crucial for the development of this type of airway disease. Airway epithelial cells and innate immune cells represent key components of the pathogenesis of chronic airway disease and are emerging targets for new therapies. In this Review, we summarize the innate immune mechanisms by which airway epithelial cells and innate immune cells regulate the development of chronic respiratory diseases. We also explain how these pathways are being targeted in the clinic to treat patients with these diseases.
Chronic lower respiratory diseases most commonly manifest as asthma or chronic obstructive pulmonary disease (COPD), and they are a leading cause of morbidity and mortality throughout the world1,2. It is widely believed that an abnormal inflammatory response to environmental agents in genetically susceptible individuals is responsible for causing this type of disease. Environmental agents that may trigger asthma or COPD include allergens, tobacco and wood smoke, and microbial pathogens. Indeed, there has been considerable progress in defining how the immune system of the lungs responds to these agents.
The conventional view has been that the adaptive immune response is crucial for the type of long-term inflammation that is required to drive chronic respiratory disease. This scheme has been particularly well developed for allergic reactions, but has also been extrapolated to explain the immune responses that are induced by non-allergic stimuli3. However, an alternative view that is gaining wider acceptance is that the innate immune system also drives chronic respiratory disease (FIG. 1). This conceptual shift raises the possibility that sentinel epithelial cells and immune cells might be essential components of pathogenesis, and might represent new targets for therapeutic intervention. A particular challenge is to explain how innate immune responses, which are traditionally viewed as being transient in nature, can drive the type of long-term immune activation that is seen in the context of chronic inflammatory disease.
Figure 1
Adaptive and innate immune responses in chronic respiratory disease
a | Environm ental stimuli — suchas respiratory viruses, allergens and/or tobacco smoke — may act on genetically susceptible individuals to lead to an altered immune response, end-organ dysfunction and chronic inflammatory disease. b | An altered adaptive immune response involves antigen-presenting cells, primarily dendritic cells (DCs), that process and present antigens to memory B cells and T cells that drive the activation of effector immune cells (such as eosinophils and mast cells). Additional T cell subsets that regulate the adaptive immune response include T helper 17 (TH17) cells, TH9 cells and regulatory T cells (not shown). Alternatively, an altered innate immune response can involve airway epithelial cells (AECs) that activate innate immune cells, such as invariant natural killer T (iNKT) cells, M2 macrophages and innate lymphoid cells (ILCs). c | Effector cells or innate immune cells then produce type 2 cytokines — for example, interleukin-4 (IL-4) and IL-13 — that act on end-organ cells, especially AECs, to produce excess mucus, and on airway smooth muscle cells (ASMCs) to manifest airway hyperreactivity, which, to varying degrees, are both characteristic of patients with asthma and chronic obstructive pulmonary disease.
Figure 2
PRR pathways in AECs leading to airway disease
Allergens such as Der-p2 derived from the house dust mite (HDM) Dermatophagoides farinae and fibrinogen cleavage products (FCPs) that are generated by proteases from the fungus Aspergillus oryzae can act as ligands for the Toll-like receptor 4 (TLR4) complex. The activation of TLR4-dependent signalling leads to an allergic response that is characterized by type 2 cytokine production. Alternatively, viral infection can induce the activation of several additional TLRs (such as TLR3, TLR7, TLR8 and TLR9) in the endosome and can also activate RIG-I-like receptors (RLRs) — such as melanoma differ entiation-associated protein 5 (MDA5) and retinoic acid-inducible gene I (RIG-I) — in the cytosol. In each case, activation leads to downstream signalling with the eventual stimulation of transcription factors in the nucleus and consequent expression of the indicated cytokines and interferon (IFN)-stimulated genes (ISGs). dsRNA, double-stranded RNA; IL, interleukin; IRF, IFN-regulatory factor; MAVS, mitochondrial antiviral signalling protein; MD2, myeloid differentiation factor 2; MYD88, myeloid differentiation primary response protein 88; NF-κB, nuclear factor-κB; ssRNA, single-stranded RNA; TNF, tumour necrosis factor; TRIF, TIR domain-containing adaptor protein inducing IFNβ.
Figure 3
Innate immune responses of AECs drive airway disease
Respiratory viral infection (which is perhaps enhanced by exposure to allergens or tobacco smoke) leads to an expansion of airway progenitor epithelial cells (APECs), which are a subset of integrin α6 (ITGA6)-expressing basal cells in humans or secretory cells expressing SCGB1A1 (also known as uteroglobin) in mice that are programmed for increased interleukin-33 (IL-33) expression. Subsequent epithelial ‘danger’ signals stimulate ATP-regulated release of IL-33 that acts on innate immune cells in the lungs — for example, type 2 innate lymphoid cells (ILC2s) and invariant natural killer T (iNKT) cells, which can interact with M2-like macrophages to stimulate IL-13 production. IL-13 then induces IL-13 receptor (IL-13R) signalling to stimulate calcium-activated chloride channel regulator 1 (CLCA1) expression and mitogen-activated protein kinase 13 (MAPK13)-dependent signalling, which activate expression of MUC5AC (which encodes mucin 5AC) and, consequently, lead to airway mucous cell activation and mucus formation. Figure modified with permission from the American Society for Clinical Investigation (REF. 16).
Figure 4
Innate immune cells in post-viral airway disease
a | Viral infection drives airway epithelial cell (AEC) release of interleukin-33 (IL-33) and the subse quent activation of invariant NKT (iNKT) cells that express an invariant Vα 14-Jα 18 T cell receptor (TCR) that recognizes glycolipids presented on CD1d molecules by lung monocytes and M2 macrophages. These signals lead to increased expression of the IL-13 receptor (IL-13R), and production of IL-13 that facilitates a positive feedback loop to amplify IL-13 production and alternative activation of monocytes and macrophages. Alternatively activated monocytes and macrophages are marked by epidermal arachidonate 12-lipoxygenase (ALOX12E), arginase 1 (ARG1), chitinase-like protein 3 (CHIL3), CHIL4, FIZZ1 (also known as resistin-like molecule-α) and matrix metalloproteinase 12 (MMP12) expression in mice, and by ALOX15, CD163, CD206, chitotriosidase 1 (CHIT1) and MMP12 expression in humans. AEC release of IL-33 may also stimulate type 2 innate lymphoid cells (ILC2s), as well as effector granulocytes (such as eosinophils, mast cells and basophils; not shown), to produce IL-13. b | Viral infection also stimulates interferon-β (IFNβ)-dependent and CD49d+ neutrophil-dependent upregulation of the high-affinity Fc receptor for IgE (FcεRI) expression on resident lung dendritic cells (DCs). In turn, FcεRI activation by viral antigens and IgE leads to the production of CC-chemokine ligand 28 (CCL28) and the recruitment of CC-chemokine receptor 10 (CCR10)-expressing IL-13-producing T helper 2 (TH2) cells to the lungs.Conclusion
It has been difficult to understand how an abnormal immune response leads to the development of chronic inflammatory disease in general, and this has also been the case for chronic respiratory diseases, such as asthma and COPD. However, ongoing progress in in vitro and in vivo models, and in corresponding human studies, suggests that the innate immune response can drive the initiation, exacerbation and progression of this type of disease in response to inhaled allergens, respiratory viruses and, most probably, other environmental stimuli. The innate immune axis seems to begin with the response of sentinel AECs that then transmit activation signals to innate immune cell populations. Recent progress provides two major insights: first, that APECs may be reprogrammed and expanded with specific cytokines (such as IL-33) to provide an ongoing susceptibility to a type 2 immune response; and second, that once activated, AECs may transmit signals to various innate immune cell populations — including NKT cells, macro phages and ILCs — that in turn produce additional cytokines (such as IL-13) to drive airway inflammation, airway hyperreactivity and excessive airway mucus production. These insights have translated into the first encouraging efforts to block these cytokine signalling pathways and treat chronic airway disease.。。。
Nonetheless, ongoing research progress already indicates that environmental stimuli (especially respiratory viral infections in synergy with allergic reactions and smoke exposure) can drive innate immune responses with consequences for acute as well as chronic respiratory disease. A better definition of the role of the AEC and innate immune cell components in chronic airway disease should enable the development of new, effective therapeutics for patients.
The role of airway epithelial cells and innate immune cells in chronic respiratory disease
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4782595/
Nat Immunol. Author manuscript; available in PMC 2015 Feb 5.
Respiratory epithelial cells orchestrate pulmonary innate immunity
Jeffrey A Whitsett1 and Theresa Alenghat2
Author information Copyright and License information Disclaimer
1Perinatal Institute, Division of Neonatology, Division of Perinatal Biology and Division of Pulmonary Biology, Cincinnati Children’s Hospital Medical Center and the University of Cincinnati College of Medicine, Cincinnati, Ohio, USA
2Division of Immunobiology, Cincinnati Children’s Hospital Medical Center and the University of Cincinnati College of Medicine, Cincinnati, Ohio, USA
Abstract
The epithelial surfaces of the lungs are in direct contact with the environment and are subjected to dynamic physical forces as airway tubes and alveoli are stretched and compressed during ventilation. Mucociliary clearance in conducting airways, reduction of surface tension in the alveoli, and maintenance of near sterility have been accommodated by the evolution of a multi-tiered innate host-defense system. The biophysical nature of pulmonary host defenses are integrated with the ability of respiratory epithelial cells to respond to and ‘instruct’ the professional immune system to protect the lungs from infection and injury.
Oxidative metabolism of cells throughout the body requires the exchange of vast quantities of oxygen and carbon dioxide across the alveolar-capillary interface in the peripheral lung. Throughout life, the dynamic process of ventilation moves millions of liters of air through the highly branched conducting airways to the alveoli, the latter lined by type I and type II epithelial cells. The gracile structure of the alveoli brings epithelial cells in close apposition to pulmonary capillaries for gas exchange. While this delivers life-requiring oxygen to the systemic circulation, particles, microbes and toxicants are also brought into the respiratory tract, where they meet a multilayered physical and chemical innate host-defense system evolved to prevent their entry into lung tissue and the circulation. Innate host defenses of the conducting airway depend on its branching structure and the multiple barriers created by layers of mucus, the tight adhesions between epithelial cells and the underlying stroma, and an abundance of fluid and antimicrobial molecules that enable mucociliary clearance. Conducting airways are the conduits whose chief role is to deliver almost completely sterile, hydrated gases to the peripheral alveoli for gas exchange (Fig. 1). In sharp anatomic contrast to the airways, the alveolar region of the lungs is a unique structural environment wherein surface tension is controlled by the careful balance of fluids and unique surface active lipids and proteins that remain stable during the expansion and compression of ventilation (Fig. 2). The anatomical structures that constitute the conducting and peripheral airways serve distinct roles in the innate defense of the lungs, and the diversity of epithelial cells lining the respiratory tract contributes in unique ways to pulmonary homeostasis.
Figure 1
Structure and function of the innate host defenses in conducting airways. Cartilaginous airways from the terminal bronchioles to the trachea are lined by a pseudostratified epithelium, whose surface is lined by ciliated and secretory cells, that together with submucosal glands, secrete mucins and other host-defense proteins into the periciliary fluids (a,b). Various transcription factors and associated proteins (b, bottom right) are selectively expressed in distinct subsets of epithelial cells lining the airways and submucosal glands. Secreted mucins (blue), such as MUC5AC and MUC5B, produced by goblet cells create a hydrated mucus gel (c,d) that binds particles and pathogens that are moved by the periciliary brush (b) up the airway for clearance from the lungs. Epithelial cells lining the airways and submucosal glands (b,d) create tight epithelial barriers and secrete a diversity of host-defense proteins that recognize microbial pathogens, which enhances the uptake and killing of those pathogens by professional cells of the immune system. The biophysical scaffolds created by the mucus gel, tight cell-cell junctions and communication among respiratory epithelial cells provide multiple barriers to infection. The secretion of fluid and mucus is coordinated with the directional beating of cilia (ultrastructure in electronmicrograph in b) mediated by Cnx43.
Figure 2
Integration of surfactant function and innate host defenses in the alveoli. Gas exchange is mediated by the close apposition of type I and type II epithelial cells to the endothelial cells of pulmonary capillaries, which creates an extensive surface area whereon environmental gases create collapsing forces at the hydrated surfaces of the alveoli (a,b). Hopx is a transcription factor selectively expressed in type I cells, and ABCA3 is a surfactant lipid transporter specific for type II epithelial cells in the alveoli (b). Antibody to smooth muscle actin (α-SMA) stains bronchiolar and vascular smooth muscle. Surface tension is diminished by pulmonary surfactant lipids and proteins secreted by type II epithelial cells (c–e) that remain stable during the dynamic compression and expansion of the lungs during ventilation. The biophysical activities of surfactant are integrated with alveolar host-defense functions that are mediated by the structural components of surfactant that have intrinsic antimicrobial activity. Tubular myelin (a,f), formed by surfactant proteins SP-A and SP-B, and lipid create a highly structured reservoir of surfactant and host-defense proteins that interact with alveolar macrophages and other cells of the immune system to bind to and remove microbial pathogens and ‘instruct’ inflammatory cells to mount appropriate host-defense responses (b). Alveolar epithelial cell and alveolar macrophages directly interact via Cnx43 channels to modify local inflammatory signals and regulate the expression of cytokines and chemokines in response to pathogens. The sizes of surfactant pools are maintained by the synthesis, secretion and reuptake of lipids and proteins by alveolar epithelial cells and by the catabolic activities of alveolar macrophages via processes regulated by GM-CSF that together maintain near sterility of the alveoli (a).
Figure 3
Signaling via PAMPs and DAMPs in respiratory epithelial cells and downstream host-defense responses. PAMPs derived from commensal microbes or respiratory pathogens and DAMPs generated from cell stress and/or death within both the conducting airways and alveoli are recognized via membrane-associated or cytosolic PRRs expressed in respiratory epithelial cells. The binding of ligands to these receptors results in the activation of epithelial cell–intrinsic signaling pathways (via MAPK, IRFs, reactive oxygen species (ROS) and NF-κB) and subsequent production of cytokines, chemokines and antimicrobial proteins that recruit and activate cells of the innate and adaptive immune systems and regulate barrier function. These same recognition pathways in epithelial cells can stimulate autophagy, phagocytosis and the clearance of necrotic cells and pathogens and thus further influence local inflammatory responses. dsRNA, double-stranded RNA.Summary
Innate defense responses of the respiratory epithelium have enabled evolutionary adaption to the constant exposure to microbial pathogens, particles and toxicants while maintaining lung function and tissue homeostasis. Respiratory epithelial cells produce a repertoire of biophysical scaffolds, host-defense molecules and barriers and communicate among themselves and with professional cells of the immune system through the production of cytokines, chemokines and DAMPs to maintain near sterility of the peripheral lungs throughout life.Respiratory epithelial cells orchestrate pulmonary innate immunity
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4318521/
Cell Death Dis. 2019 Jun 5;10(6):442. doi: 10.1038/s41419-019-1684-0.
H7N9 influenza A virus activation of necroptosis in human monocytes links innate and adaptive immune responses.
Lee ACY1, Zhang AJX1,2,3,4, Chu H1,2,3,4, Li C1, Zhu H1, Mak WWN1, Chen Y1, Kok KH1,2,3,4, To KKW1,2,3,4, Yuen KY5,6,7,8.
Author information1
Department of Microbiology, The University of Hong Kong, Hong Kong, China.
2
State Key Laboratory of Emerging Infectious Diseases, Hong Kong, China.
3
Carol Yu Centre for infection, Hong Kong, China.
4
Research Centre of Infection and Immunology, The University of Hong Kong, Hong Kong, China.
5
Department of Microbiology, The University of Hong Kong, Hong Kong, China. kyyuen@hku.hk.
6
State Key Laboratory of Emerging Infectious Diseases, Hong Kong, China. kyyufen@hku.hk.
7
Carol Yu Centre for infection, Hong Kong, China. kyyuen@hku.hk.
8
Research Centre of Infection and Immunology, The University of Hong Kong, Hong Kong, China. kyyuen@hku.hk.
Abstract
We previously demonstrated that avian influenza A H7N9 virus preferentially infected CD14+ monocyte in human peripheral blood mononuclear cells (PBMCs), which led to apoptosis. To better understand H7N9 pathogenesis in relation to monocyte cell death, we showed here that extensive phosphorylation of mixed lineage kinase domain-like (MLKL) protein occurred concurrently with the activation of caspases-8, -9 and -3 in H7N9-infected monocytes at 6 h post infection (hpi), indicating that apoptosis and necroptosis pathways were simultaneously activated. The apoptotic morphology was readily observed in H7N9-infected monocytes with transmission electron microscopy (TEM), while the pan-caspase inhibitor, IDN6556 (IDN), accelerated cell death through necroptosis as evidenced by the increased level of pMLKL accompanied with cell swelling and plasma membrane rupture. Most importantly, H7N9-induced cell death could only be stopped by the combined treatment of IDN and necrosulfonamide (NSA), a pMLKL membrane translocation inhibitor, but not by individual inhibition of caspase or RIPK3. Our data further showed that activation of apoptosis and necroptosis pathways in monocytes differentially contributed to the immune response of monocytes upon H7N9 infection. Specifically, caspase inhibition significantly enhanced, while RIPK3 inhibition reduced the early expression of type I interferons and cytokine/chemokines in H7N9-infected monocytes. Moreover, culture supernatants from IDN-treated H7N9-infected monocyte promoted the expression of co-stimulatory molecule CD80, CD83 and CD86 on freshly isolated monocytes and monocyte-derived dendritic cells (MDCs) and enhanced the capacity of MDCs to induce CD3+ T-cell proliferation in vitro. In contrast, these immune stimulatory effects were abrogated by using culture supernatants from H7N9-infected monocyte with RIPK3 inhibition. In conclusion, our findings indicated that H7N9 infection activated both apoptosis and necroptosis in monocytes. An intact RIPK3 activity is required for upregulation of innate immune responses, while caspase activation suppresses the immune response.
PMID: 31165725 PMCID: PMC6549191 DOI: 10.1038/s41419-019-1684-0H7N9 influenza A virus activation of necroptosis in human monocytes links innate and adaptive immune responses. - PubMed - NCBI
https://www.ncbi.nlm.nih.gov/pubmed/31165725
Targeted Infection of Endothelial Cells by Avian Influenza ...
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC112334
Thus, the polarity of virus maturation in endothelial cells may be another factor promoting spread of infection via the blood while hindering infection of subendothelial tissues. DISCUSSION We have found in this study that FPV shows strict endotheliotropism when infecting 11-day-old chick embryos.
Cited by: 109
Publish Year: 2000
Author: Anke Feldmann, Martin K.-H. Schäfer, Wolfgang Garten, Hans-Dieter Klenk
Roles for Endothelial Cells in Dengue Virus Infection
https://www.hindawi.com/journals/av/2012/840654
Cell lines derived from endothelial and epithelial cell fusions are not representative of primary ECs and the ECV304 endothelial cell line has been shown to be bladder carcinoma and not endothelial in nature . However, early passage primary human ECs permit investigation of dengue virus infection that approximates the human endothelium for ...
Cited by: 58
Publish Year: 2012
Author: Nadine A. Dalrymple, Erich R. Mackow
American Society for Microbiology Journals, 2012Alveolar Epithelial Cells Are Critical in Protection of the Respiratory Tract by Secretion of Factors Able To Modulate the Activity of Pulmonary Macrophages and Directly Control Bacterial Growth
Olga D. Chuquimia, Dagbjort H. Petursdottir, Natalia Periolo, Carmen Fernández
J. L. Flynn, Editor
- Department of Immunology, Wenner-Gren Institute, Stockholm University, Stockholm, Sweden
ABSTRACT
The respiratory epithelium is a physical and functional barrier actively involved in the clearance of environmental agents. The alveolar compartment is lined with membranous pneumocytes, known as type I alveolar epithelial cells (AEC I), and granular pneumocytes, type II alveolar epithelial cells (AEC II). AEC II are responsible for epithelial reparation upon injury and ion transport and are very active immunologically, contributing to lung defense by secreting antimicrobial factors. AEC II also secrete a broad variety of factors, such as cytokines and chemokines, involved in activation and differentiation of immune cells and are able to present antigen to specific T cells.Another cell type important in lung defense is the pulmonary macrophage (PuM). Considering the architecture of the alveoli, a good communication between the external and the internal compartments is crucial to mount effective responses.
Our hypothesis is that being in the interface, AEC may play an important role in transmitting signals from the external to the internal compartment and in modulating the activity of PuM. For this, we collected supernatants from AEC unstimulated or stimulated in vitro with lipopolysaccharide (LPS). These AEC-conditioned media were used in various setups to test for the effects on a number of macrophage functions: (i) migration, (ii) phagocytosis and intracellular control of bacterial growth, and (iii) phenotypic changes and morphology. Finally, we tested the direct effect of AEC-conditioned media on bacterial growth.
AEC II are cuboidal cells that constitute around 15% of total lung cells and cover about 7% of the total alveolar surface. AEC II are responsible for epithelium reparation upon injury and ion transport. AEC II contribute also to lung defense by secreting antimicrobial products such as complement, lysozyme, and surfactant proteins (SP).
We found that AEC-secreted factors had a dual effect, on one hand controlling bacterial growth and on the other hand increasing macrophage activity.
Alveolar Epithelial Cells Are Critical in Protection of the Respiratory Tract by Secretion of Factors Able To Modulate the Activity of Pulmonary Macrophages and Directly Control Bacterial Growth | Infection and Immunity
https://iai.asm.org/content/81/1/381/article-info
Alveolar Type II Epithelial Cells Contribute to the Anti-Influenza A Virus Response in the Lung by Integrating Pathogen- and Microenvironment-Derived Signals
S. Stegemann-Koniszewski, Andreas Jeron, Marcus Gereke, Robert Geffers, Andrea Kröger, Matthias Gunzer, Dunja Bruder
Michael G. Katze, EditoraImmune Regulation, Helmholtz Centre for Infection Research, Braunschweig, Germany
bInfection Immunology, Institute of Medical Microbiology, Infection Control and Prevention, Otto-von-Guericke University, Magdeburg, Germany
cGenome Analytics, Helmholtz Centre for Infection Research, Braunschweig, Germany
dInnate Immunity and Infection, Helmholtz Centre for Infection Research, Braunschweig, Germany
eMolecular Microbiology, Institute of Medical Microbiology, Infection Control and Prevention, Otto-von-Guericke University, Magdeburg, Germanye
fInstitute of Experimental Immunology and Imaging, University of Duisburg-Essen, Essen, Germany
ABSTRACT
Influenza A virus (IAV) periodically causes substantial morbidity and mortality in the human population. In the lower lung, the primary targets for IAV replication are type II alveolar epithelial cells (AECII), which are increasingly recognized for their immunological potential.So far, little is known about their reaction to IAV and their contribution to respiratory antiviral immunity in vivo. Therefore, we characterized the AECII response during early IAV infection by analyzing transcriptional regulation in cells sorted from the lungs of infected mice.
We detected rapid and extensive regulation of gene expression in AECII following in vivo IAV infection. The comparison to transcriptional regulation in lung tissue revealed a strong contribution of AECII to the respiratory response. IAV infection triggered the expression of a plethora of antiviral factors and immune mediators in AECII with a high prevalence for interferon-stimulated genes. Functional pathway analyses revealed high activity in pathogen recognition, immune cell recruitment, and antigen presentation. Ultimately, our analyses of transcriptional regulation in AECII and lung tissue as well as interferon I/III levels and cell recruitment indicated AECII to integrate signals provided by direct pathogen recognition and surrounding cells.
Ex vivo analysis of AECII proved a powerful tool to increase our understanding of their role in respiratory immune responses, and our results clearly show that AECII need to be considered a part of the surveillance and effector system of the lower respiratory tract.
IMPORTANCE In order to confront the health hazard posed by IAV, we need to complete our understanding of its pathogenesis. AECII are primary targets for IAV replication in the lung, and while we are beginning to understand their importance for respiratory immunity, the in vivo AECII response during IAV infection has not been analyzed. In contrast to studies addressing the response of AECII infected with IAV ex vivo, we have performed detailed gene transcriptional profiling of AECII isolated from the lungs of infected mice. Thereby, we have identified an exceptionally rapid and versatile response to IAV infection that is shaped by pathogen-derived as well as microenvironment-derived signals and aims at the induction of antiviral measures and the recruitment and activation of immune cells. In conclusion, our study presents AECII as active players in antiviral defense in vivo that need to be considered part of the sentinel and effector immune system of the lung.
FIG 6
Integrative model of the contribution of AECII to lower respiratory tract anti-IAV responses. The analysis of transcriptional regulation in AECII isolated from infected mice revealed their rapid and versatile immunological response to IAV infection in vivo. Based on our results, we propose that AECII integrate signals from a direct interaction with the pathogen with signals provided through cytokines released by additional sentinel cells in order to mount this response.INTRODUCTION
Influenza A virus (IAV) still poses a serious threat to human health, and a detailed understanding of IAV pathogenesis is essential to adequately confront this hazard. IAV infections are primarily restricted to the respiratory tract, where epithelial cells, alveolar macrophages (AM), and dendritic cells (DC) trigger the first innate responses (1). IAV bears ligands for several pathogen recognition receptors (PRR), and the main triggered PRR are Toll-like receptor 3 (TLR3) and TLR7 as well as RIG-I, MDA5, and the NLRP3 inflammasome (1). These are engaged in the antiviral response in a cell-type-specific manner (2, 3). Via partly redundant signaling pathways, PRR ligation leads to the activation of effector mechanisms comprised of type I/III interferons (IFNs), inflammatory mediators, antimicrobial effectors, and signals inducing adaptive immunity. In general, viral infections are marked by the strong release of type I interferons. These trigger the expression of a multitude of interferon-stimulated genes (ISG) through the ubiquitously expressed IFN-α/β receptor (IFNAR) (2). ISG expression is also induced through IFN-λ (IFN III), which is released during IAV infection and is sensed through the interleukin-28 (IL-28) receptor α (IL-28Rα) primarily expressed by epithelial cells of the respiratory tract and gut (4).
In the lower respiratory tract, the lining epithelium is comprised of alveolar type I and type II epithelial cells (AECI and AECII, respectively), and at this site, AECII are the main target cells for IAV replication (5, 6). AECII cover about 5% of the alveolar surface, while they comprise about 60% of the alveolar lining cells and 15% of the parenchymal cells (7). Until recently, the secretion of surfactant, the maintenance of the mechanical barrier, and the provision of constitutive antimicrobial defense were conceived as their main functions (7, 8). Beyond these, we are only beginning to understand the potential of AECII to regulate respiratory immune responses in autoimmunity and infection (9–12).
Since AECII are primary targets for viral replication in the lower lung and actively contribute to pulmonary immunity, it is likely that they influence efficient host responses directed at IAV. A number of studies have addressed the response of AECII to IAV in vitro and showed them to express functional PRR and to produce cytokines and chemokines (13–17). The nature and the relevance of the AECII response to IAV, however, lack ultimate clarification, as these studies were performed using cell lines or primary cells infected in culture. Little is known about the AECII response to IAV infection in vivo and how this contributes to the respiratory immune reaction. To overcome these limitations, we characterized the in vivo response of AECII to IAV infection by analyzing primary AECII from the lungs of infected mice.Alveolar Type II Epithelial Cells Contribute to the Anti-Influenza A Virus Response in the Lung by Integrating Pathogen- and Microenvironment-Derived Signals | mBio
https://mbio.asm.org/content/7/3/e00276-16?utm_source=TrendMDmBio&utm_medium=TrendMDmBio&utm_campaign=trendmdalljournals_0
Cellular Response to Infection | Spotlight
Innate Immune Response to Influenza Virus at Single-Cell Resolution in Human Epithelial Cells Revealed Paracrine Induction of Interferon Lambda 1
Irene Ramos, Gregory Smith, Frederique Ruf-Zamojski, Carles Martínez-Romero, Miguel Fribourg, Edwin A. Carbajal, Boris M. Hartmann, Venugopalan D. Nair, Nada Marjanovic, Paula L. Monteagudo, Veronica A. DeJesus, Tinaye Mutetwa, Michel Zamojski, Gene S. Tan, Ciriyam Jayaprakash, Elena Zaslavsky, Randy A. Albrecht, Stuart C. Sealfon, Adolfo García-Sastre, Ana Fernandez-Sesma
Bryan R. G. Williams, Editor
DOI: 10.1128/JVI.00559-19
- aDepartment of Microbiology, Icahn School of Medicine at Mount Sinai, New York, New York, USA
- bDepartment of Neurology, Center for Advanced Research on Diagnostic Assays, Icahn School of Medicine at Mount Sinai, New York, New York, USA
- cDepartment of Medicine, Icahn School of Medicine at Mount Sinai, New York, New York, USA
- dThe Graduate School of Biomedical Sciences, Icahn School of Medicine at Mount Sinai, New York, New York, USA
- eGlobal Health and Emerging Pathogens Institute, Icahn School of Medicine at Mount Sinai, New York, New York, USA
- fInfectious Diseases, J. Craig Venter Institute, La Jolla, California, USA
- gDepartment of Medicine, University of California San Diego, La Jolla, California, USA
- hDepartment of Physics, The Ohio State University, Columbus, Ohio, USA
ABSTRACT
Early interactions of influenza A virus (IAV) with respiratory epithelium might determine the outcome of infection.The study of global cellular innate immune responses often masks multiple aspects of the mechanisms by which populations of cells work as organized and heterogeneous systems to defeat virus infection, and how the virus counteracts these systems.
In this study, we experimentally dissected the dynamics of IAV and human epithelial respiratory cell interaction during early infection at the single-cell level. We found that the number of viruses infecting a cell (multiplicity of infection [MOI]) influences the magnitude of virus antagonism of the host innate antiviral response. Infections performed at high MOIs resulted in increased viral gene expression per cell and stronger antagonist effect than infections at low MOIs. In addition, single-cell patterns of expression of interferons (IFN) and IFN-stimulated genes (ISGs) provided important insights into the contributions of the infected and bystander cells to the innate immune responses during infection. Specifically, the expression of multiple ISGs was lower in infected than in bystander cells. In contrast with other IFNs, IFN lambda 1 (IFNL1) showed a widespread pattern of expression, suggesting a different cell-to-cell propagation mechanism more reliant on paracrine signaling. Finally, we measured the dynamics of the antiviral response in primary human epithelial cells, which highlighted the importance of early innate immune responses at inhibiting virus spread.
IMPORTANCE Influenza A virus (IAV) is a respiratory pathogen of high importance to public health. Annual epidemics of seasonal IAV infections in humans are a significant public health and economic burden. IAV also causes sporadic pandemics, which can have devastating effects. The main target cells for IAV replication are epithelial cells in the respiratory epithelium. The cellular innate immune responses induced in these cells upon infection are critical for defense against the virus, and therefore, it is important to understand the complex interactions between the virus and the host cells. In this study, we investigated the innate immune response to IAV in the respiratory epithelium at the single-cell level, providing a better understanding on how a population of epithelial cells functions as a complex system to orchestrate the response to virus infection and how the virus counteracts this system.
Early innate immune responses restrict virus replication in IAV-infected human primary respiratory epithelial cells.Our results show that IAV infection at a low MOI results in induction of antiviral ISGs in bystander cells that potentially protect them from viral infection. In order to address the magnitude and timing of the antiviral response induced in bystander cells during IAV infection at a low MOI using primary human respiratory epithelial cells, we established a model of staggered IAV H1N1/H3N2 coinfections in normal human bronchial epithelial (NHBE) cells in an air-liquid interface (1). Using two similar, but distinguishable, viruses to infect cell monolayers at different times allows the analysis of how the antiviral response induced after the first infection affects subsequent infection by the second virus. The design of the experimental protocol is depicted in Fig. 8A. First, we infected NHBE cells with the H1N1 virus A/California/04/2009 at an MOI of 0.05, and then we characterized profiles of gene expression and virus replication throughout a 48-h time course. Using plaque assays (Fig. 8B), we established that the initial release of H1N1 infectious particles was detected at 12 hpi, peaked at 24 to 36 hpi, and declined after 48 hpi. We next analyzed the kinetics of expression of a comprehensive panel of innate immunity-associated genes by quantitative reverse transcriptase PCR (qRT-PCR). Notably, we detected early upregulation of IFNB1, at around 4 hpi, as well as upregulation of IFNL1, IFNL2, and IFNL3, while changes in IFNA expression remained undetected (Fig. 8C). Interestingly, ISGs, including IFIT2, CXCL10, RIGI, and Mx1, were also detected very early after infection (4 to 12 hpi [Fig. 8D]). Therefore, these results demonstrate an early expression of type I and type III IFN and ISGs during IAV infection in epithelial cells.
FIG 8
Open in new tabDownload powerpoint
FIG 8
Analysis of the functional antiviral response induced by IAV in differentiated NHBE cells using a H1N1/H3N2 coinfection assay. (A) Scheme of the experimental setup and time line. (B) Replication profile of H1N1 (Cal09) infection of NHBE cells. (C and D) Expression of type I and III IFN and ISGs in NHBE cells in cultures infected with the H1N1 IAV or mock infected. (E) Impact of the innate immune response induced by H1N1 IAV infection on the replication of the H3N2 virus. Averages of biological triplicates ± SEMs are shown. Two-way analysis of variance (ANOVA) and Tukey’s multiple-comparison test were used. Adjusted P values are shown as follows: *, <0.05; **, <0.01; and ***, <0.001. (F) Immunofluorescence assay of H1N1-infected NHBE cells, showing a time course analysis of the progression of viral infection.
In the same experiment, additional samples were first infected with H1N1 virus, and then the H3N2 virus A/Wyoming/03/2003 was added at an MOI of 0.05 at selected times points post-H1N1 infection in the presence of the H1 neutralizing antibody 7B2 (Fig. 8A), which is specific for the head region of the A/California/04/2009 isolate and restricts its subsequent rounds of infection. Washes from the apical chamber were collected to assess viral replication at 1, 12, 24, and 48 h after H3N2 infection. The viral titers (PFU) measured after H3N2 infection correspond with H3N2 replication only, since the anti-H1 antibody restricts infections with H1N1 viruses. We used changes in those H3N2 titers as an indicator of the functional antiviral response induced by the previously added H1N1 virus. As both H1N1 and H3N2 viruses were added at a low MOI, numbers of coinfections were negligible. Control experiments demonstrated that the neutralizing antibody 7B2 inhibited H1N1 replication as measured by plaque assay and immunostaining (data not shown). Thus, in this experiment we measured levels of H3N2 replication in bystander cells exposed to paracrine effects from H1N1-infected cells. Analysis of the impact of the innate immune response induced by the initial H1N1 IAV infection on the replication of the subsequently added H3N2 virus showed that adding the H3N2 virus at 2 or 4 hpi resulted in a replication profile very similar to that obtained with the H3N2 single infection. Interestingly, we found a significant decrease in the H3N2 replication titers (48 h post-H3N2 infection) when this virus was added as early as 8 and 12 h post-H1N1 IAV infection. As expected, we observed a greater reduction of the H3N2 IAV replication as the time of addition post-H1N1 IAV infection increased to 24 h or 48 h. To confirm that after infection with the H1N1 virus there was still a sufficiently large number of uninfected cells that could be potentially infected by the H3N2 virus, we performed immunofluorescence staining of NHBE cells infected by H1N1 under the same conditions. Visualization and quantification of complete wells indicated the presence of 1.23% ± 1.32%, 3.2% ± 1.37%, and 6.01% ± 3.00% infected cells at 12, 24, and 48 hpi, respectively (Fig. 8F), indicating that most cells were available for infection upon exposure to the second virus. Therefore, these data indicate that early IFN responses elicited during initial viral infection are effective as early as 8 to 12 hpi and are critical for a functional antiviral response in bystander cells.
DISCUSSION
The innate immune response is one of the first mechanisms of host defense against viral infections. In order to successfully establish infection in humans, viruses such as IAV have developed strategies to counteract this system. While extensive research on the genes involved in antiviral responses and on IAV antagonism has been performed, the global virus-host interaction and its consequences at the individual cell level are still not well understood. In this study, we performed a deep analysis of these interactions and their functional consequences in respiratory epithelial cells, which are main targets of IAV replication.
Our analysis of the dynamics of NS1 expression showed an MOI-dependent expression of this important innate immune antagonist protein in infected cells. Similarly, we found that the levels of all IAV genes per infected cell were more elevated when cells were exposed to a high MOI than to a low MOI, most likely due to differences in the numbers of cells being initially infected with multiple virions. Under physiological conditions, the first round of infection of the respiratory epithelia by IAV would be mediated by a few virus particles, so probably infected host cells receive only one infectious virus. This is consistent with the very low number of viruses shown to be responsible for initation of infection in ferrets (53) and humans (54). However, as infection progresses, infection of neighboring cells is likely mediated by more than one virus originating from the infected cells, with lower numbers of viruses reaching cells farther away from the initially infected cells. Thus, we expect to find both single-virus- and multiple-virus-infected cells during human IAV infections. Interestingly, as found by a correlation analysis of the viral genes versus a panel of innate immune genes, NS shows a consistent negative correlation with the transcription of most of the innate immune genes. This negative correlation is clearly more enhanced in cells infected with multiple viruses (high MOI) than in those infected with single viruses (low MOI), suggesting that the number of virions that infect each individual cell has important consequences for the ability of the virus to counteract the innate immune response in that cell (see model in Fig. 9). Therefore, our results suggest that viral IFN antagonism might be less effective during the initial stages of infection in the context of humans infected with a virus, when cells are impacted by single virus particles. During viral infection, the concentration of viral particles released grows exponentially, reaching levels up to 105 to 107 50% tissue culture infective doses (TCID50)/ml (55, 56). At this level of replication in the respiratory tract tissue, it is highly probable that many cells get infected with multiple viruses, which would enhance the ability of IAV to suppress the innate immune response in vivo and at the same time favor the reassortment of IAV genes, an important evolutionary force for IAV.
FIG 9
Open in new tabDownload powerpoint
FIG 9
Model of the interplay between IAV and innate immunity in epithelial cells during infection with individual virus particles (left) versus multiple virus particles per cell (right). This illustration was created with BioRender (https://biorender.com).
NS1 has multiple functions to antagonize the cellular response. One of the best characterized is its ability to inhibit virus sensing by RIGI, either by sequestering dsRNA or by direct interaction with RIGI or TRIM25 (19, 57–59). Related to this, we found IFNB1, all type III IFNs, and other cytokines and chemokines among the genes that correlated negatively with the levels of NS at the high MOI. However, we found that gene expression was affected at a general level, suggesting a global inhibition of the transcription of the innate immune genes. Also, when we compared the gene expression profiles in infected versus bystander cells, we found a group of genes, presumably induced in a paracrine manner, that were expressed at lower levels in infected than in bystander cells, probably due to virus transcriptional antagonism. While a combination of viral factors could be responsible for this global shutoff of host transcription, our data suggest that NS1 might be associated with this phenomenon. There is evidence in the literature that IAV infection results in reduced host transcription and mRNA processing by inhibiting the host RNA polymerase II (Pol II) function (60, 61). Related to this, a recent study found that several strains of IAV elicit global deregulation of Poll II transcription termination by impairing 3′-end cleavage and termination, and this effect was dependent on NS1 expression (62). These studies could partially explain the global negative correlation that we found between viral genes and cellular genes. NS1 interactome studies have identified multiple additional interactions of NS1 with nuclear proteins (63, 64), which could also have a global effect on cellular transcription. In addition, another study that used a combination of modeling and experimental research found that ISGs are specifically suppressed in IAV-infected dendritic cells through the inhibition of multiple transcription factors (65). The second viral gene that showed the strongest negative correlation with host genes in our analysis was PA. Interestingly, in addition to encoding one subunit of the IAV polymerase, it has a second open reading frame accessed by ribosomal frameshifting that results in the expression of the protein known as PA-X (46). PA-X inhibits transcription of cellular genes by selectively targeting host Pol II transcribed mRNAs (66). Therefore, it is possible that the global negative correlation is a consequence of the expression of NS1 and PA-X in infected cells. Further investigation will determine if this effect is similar for other IAV strains or if it is strain specific, which will shed light into possible unknown mechanisms for virus-mediated host transcription inhibition.
The interaction between virus infection and the host response was further characterized in our study by comparing the gene expression profiles in infected and in bystander cells. Several genes, including the IFIT2, OASL and RSAD2 (also known as viperin) genes, with known antiviral activity (67–70), and IFIH1 or MDA5, which contributes to viral sensing (71), showed higher expression in infected versus bystander cells, suggesting that these ISGs, in addition to being inducible by type I or III IFN, have also an autocrine component of activation. Indeed, the induction of some ISGs was detected as early as 4 hpi (Fig. 2), with such an early upregulation consistent with direct induction by viral infection. This is in contrast to the highly accepted concept that type I or type III IFN signaling is necessary for the induction of antiviral genes (57, 72), with a few exceptions such as ISG15 and CXCL10 (73–75). The well-described mechanism of activation of transcription of ISGs upon type I/III IFN induction involves assembly and translocation to the nucleus of ISGF3, which binds to the DNA sequences in the promoters of ISGs known as interferon-stimulated response elements (ISRE) (10). Interestingly, Daly and Reich found two complexes in addition to ISGF3, interferon responsive factor 3 (IRF3) and the transcriptional coactivator CREB-binding protein (CBP)/p300, which could recognize ISRE sequences and activate transcription upon dsRNA stimulation in the absence of IFN (73, 76). The abilities of these transcription factors to interact with ISRE differ among various ISGs, due to variations in their DNA sequences. A later study identified IFIT2, ISG15, IFIT3, IFIT1, and GBP1 as IRF3 target genes (77). With the exception of these highly informative studies, literature in this specific topic is scarce. Therefore, direct induction of ISGs in infected cells might be an important player in promoting effective early antiviral responses.
The analysis of the distribution of expression of type I and III IFNs also provided very interesting observations. First, consistent with previous reports (78–80), only a small population of infected cells produced high levels of IFN. NS1 protein could differentially inhibit the IFN response across the infected cells in the culture according to the NS1 intracellular levels, which could be determined by the number of virions infecting the cells. Related to this, at the MOI of 2 we found that the cluster of cells expressing IFN at high levels (Fig. 6A) presented lower levels of the NS gene than the rest of the infected cells. Another possibility is the presence of low levels of defective interfering (DI) particles in the virus stock, which are known to be strong inducers of IFN (81). While virus stocks were carefully prepared to avoid the presence of DI particles, some presence of them cannot be ruled out. However, if a large deletion is present in one segment of a virus, mRNA for the proteins that segment encodes would not be successfully transcribed in the cell infected by that virus. Related to this, we found that at MOIs of 2 and 0.2, 100% and 99.9% of the infected cells, respectively, presented all the different RNA transcripts from the virus, suggesting a low probability for the presence of DI particles in the virus stock. Interestingly, a recent study that used an elegant approach to perform combined cellular and viral single-cell RNA sequencing in IAV-infected A549 cells found that increased proportions of viral defects or mutations, and specifically defects or deletions in NS, were highly associated with increased expression of IFN among infected cells (80).
Our single-cell sequencing data also showed that while the expression of most of the type I or III IFN is restricted to that small cell population, IFNL1 is more widespread, since it is expressed by most of the infected cells and also in the bystander cells, albeit to lower levels. The mechanism for IFNL1 induction in bystander cells is consistent with a paracrine response involving host factors secreted from virus-infected cells. Interestingly, Ank et al. (82) found that HepG2 cells (a human liver carcinoma cell line) showed upregulated expression of IFNL1 and IFNL2/3 upon treatment with either type I or type III IFN. However, in our study we did not detect induction of IFNL1 by those cytokines in A549 cells. These different results could be a consequence of the different cell types used in their study or of limits of detection. While human type I and type III IFNs are known to be induced by direct virus infection by similar transcription factors, their promoters are significantly different. There are 13 subtypes of human IFNA genes, and they are regulated by IRF3 and IRF7, which bind a cluster of transcription sites in the promoters with different affinities (83, 84). However, the regulation of expression of IFNB1 is more tightly regulated by the coordinated binding of multiple complexes of transcription factors, specifically IRF3/IRF7, nuclear factor κB (NF-κB), and activator protein 1 (AP1), to the positive regulatory domains (PRD) of the enhanceosome (85). The type III IFN has been more recently described, given its later discovery (86, 87). Human IFNL2 and IFNL3 are almost identical and have highly similar promoters (86, 87). IFNL4 is expressed in only a fraction of the human population, due to a widespread genetic polymorphism introducing a 5′-proximal frameshift (88). All IFNL promoters have binding sites for NF-κB and for IRF3/7, with some differences among them. In general, the literature suggests that IFNL promoters are more flexible than the type I IFN promoter (8), which agrees with the broader distribution of type III IFN found in our study, and that NF-κB plays an important role in its activation. IFNL1 regulation is very divergent from IFNL2/3 (13). IFNL1 has a distal cluster of NF-κB binding sites upstream of the promoter, and engagement of these sites by NF-κB transcription factors is enough to activate its expression (8, 89). Additionally, IFNL1 promoter can be activated by IRF and NF-κB factors independently through the separated elements, although engagement of both is necessary for the highest levels of expression (89). Therefore, the paracrine induction of IFNL1 could be due to its promoter organization, which is unique among the rest of the type I or type III IFNs. Alternatively, additional binding sites for transcription factors yet unidentified could be present in the IFNL1 promoter, which could contribute to the unexpected paracrine induction of this gene. The broad expression of IFNL1 suggests a possible mechanism of amplification of the antiviral response in IFNLR1-IL10R2-expressing tissues, such as the respiratory epithelium, during IAV infections.
Our study also highlights the appropriateness of single-cell transcriptome analyses to better understand the processes governing virus-host interactions. In this study, this technology allowed for the identification of patterns of expression of IFN genes and ISGs in infected cultures, suggesting that the ability of a cell to secrete IFN and the degree of their antiviral capacity are associated with multiple factors, such as their infection state and the levels of the expression of viral genes. These issues cannot be addressed by the use of bulk analysis. Given the recent development of this type of technology, only a few studies have been reported to date applying them to IAV infection. Two recent in vitro studies focused on the heterogeneity of viral transcripts during IAV infection in epithelial cells and on the characterization of virus species triggering the immune response (43, 80). In these studies, very low induction of the expression of innate immune genes was found, probably due to the use of a low MOI, which minimized the presence of coinfections as required by the type of questions addressed. An interesting in vivo study also addressed similar questions but focused mostly on the heterogeneity of infection and innate immune responses across multiple cell types (45).
We also find that early innate immune responses developed after IAV infection at low MOIs are efficient at inhibiting subsequent infection of bystander cells. If the virus overcomes this first barrier and propagates in the tissue by multiple viruses infecting the same cells, the virus would more efficiently block the innate immune response in infected cells and hypothetically could replicate more efficiently as infection progresses. Therefore, this report provides insights into how the dynamics of the interplay of IAV immune antagonism and innate immune response can change as the number of viruses infecting single cells increases in the respiratory epithilium after the early stages of infection.
Copyright © 2019 Ramos et al.
Innate Immune Response to Influenza Virus at Single-Cell Resolution in Human Epithelial Cells Revealed Paracrine Induction of Interferon Lambda 1 | Journal of Virology
https://jvi.asm.org/content/93/20/e00559-19.full
Multiplicity of infection
Jan 13, 2011 · Multiplicity of infection (MOI) is a frequently used term in virology which refers to the number of virions that are added per cell during infection. If one million virions are added to one million cells, the MOI is one. If ten million virions are added, the MOI is ten. Add 100,000 virions, and the MOI
IFNL1 - Interferon lambda-1 precursor - Homo sapiens ...
Cytokine with antiviral, antitumour and immunomodulatory activities. Plays a critical role in the antiviral host defense, predominantly in the epithelial tissues. Acts as a ligand for the heterodimeric class II cytokine receptor composed of IL10RB and IFNLR1, and receptor engagement leads to the activation of the JAK/STAT signaling pathway resulting in the expression of IFN-stimulated genes ...
Cytokine with antiviral, antitumour and immunomodulatory activities. Plays a critical role in the antiviral host defense, predominantly in the epithelial tissues. Acts as a ligand for the heterodimeric class II cytokine receptor composed of IL10RB and IFNLR1, and receptor engagement leads to the activation of the JAK/STAT signaling pathway resulting in the expression of IFN-stimulated genes (ISG), which mediate the antiviral state. Has a restricted receptor distribution and therefore restricted targets: is primarily active in epithelial cells and this cell type-selective action is because of the epithelial cell-specific expression of its receptor IFNLR1. Exerts an immunomodulatory effect by up-regulating MHC class I antigen expression.
J Clin Invest. 1994 Aug; 94(2): 731–740.
Alveolar type II epithelial cells produce interleukin-6 in vitro and in vivo. Regulation by alveolar macrophage secretory products.
B Crestani, P Cornillet, M Dehoux, C Rolland, M Guenounou, and M Aubier
Institut National de la Santé et de la Recherche Médicale INSERM U 408, Faculté Xavier Bichat, Paris, France
Abstract
The aims of this study were (a) to determine if rat alveolar type II (ATII) cells and human pulmonary epithelial-derived cells (A549 cell line) could generate IL-6 in vitro, (b) to characterize the cytokine regulation of IL-6 gene and protein expression in these cells, and (c) to detect the in vivo expression of immunoreactive IL-6 by human ATII cells.Rat ATII cells in primary culture secreted bioactive IL-6 and immunostained with an anti-IL-6 antiserum.
Spontaneous IL-6 secretion by rat ATII cells amounted to 5,690 +/- 770 pg/ml/10(6) cells (n = 12) and was fivefold higher than spontaneous rat alveolar macrophages IL-6 secretion (1,052 +/- 286 pg/ml/10(6) cells, n = 8, P = 0.001).
Rat alveolar macrophage conditioned media (CM) increased IL-6 secretion by rat ATII cells through the effect of IL-1 and TNF. IL-6 gene expression and IL-6 secretion by A549 cells was induced by IL-1 beta, TNF alpha, and by human alveolar macrophages and THP1 cells CM. Induction was abolished when CM were preincubated with anti-IL-1 beta and anti-TNF alpha antibody.
The combination of IFN gamma and LPS induced the expression of IL-6 mRNA by A549 cells whereas LPS alone had no effect. Immunohistochemical staining evidenced the expression of immunoreactive IL-6 by hyperplastic ATII cells in fibrotic human lung, a condition in which alveolar macrophages are known to be activated.
ATII cells in normal human lung did not express immunoreactive IL-6.
Our findings demonstrate that ATII cells may be an important source of IL-6 in the alveolar space thereby participating to the regulation of the intra-alveolar immune response.
Alveolar type II epithelial cells produce interleukin-6 in vitro and in vivo. Regulation by alveolar macrophage secretory products.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC296153/
Int J Biol Sci 2012; 8(9):1227-1236. doi:10.7150/ijbs.4666
Targeting Interleukin-6: All the Way to Treat Autoimmune and Inflammatory Diseases
Toshio Tanaka1,2,3, Tadamitsu Kishimoto4 Corresponding address
1. Department of Clinical Application of Biologics, Osaka University Graduate School of Medicine, Osaka University, Osaka, Japan;
2. Department of Respiratory Medicine, Allergy and Rheumatic Diseases, Osaka University Graduate School of Medicine, Osaka University, Osaka, Japan;
3. Department of Immunopathology, Immunology Frontier Research Center, Osaka University, Osaka, Japan;
4. Laboratory of Immunoregulation, Immunology Frontier Research Center, Osaka University, Osaka, Japan.
Astract
Interleukin (IL)-6, a cytokine featuring redundancy and pleiotropic activity, contributes to host defense against acute environmental stress, while dysregulated persistent IL-6 production has been demonstrated to play a pathological role in various autoimmune and chronic inflammatory diseases. Targeting IL-6 is thus a rational approach to the treatment of these diseases. Indeed, clinical trials of tocilizumab, a humanized anti-IL-6 receptor antibody have verified its efficacy and tolerable safety for patients with rheumatoid arthritis, Castleman's disease and systemic juvenile idiopathic arthritis, resulting in approval of this innovative biologic for treatment of these diseases. Moreover, a considerable number of case reports and pilot studies of off-label use of tocilizumab point to the beneficial effects of tocilizumab for a variety of other phenotypically different autoimmune and chronic inflammatory diseases. Elucidation of the source of IL-6 and of mechanisms through which IL-6 production is dysregulated can thus be expected to lead to clarification of the pathogenesis of various diseases.
Keywords: interleukin-6, a humanized anti-interleukin-6 receptor antibody, tocilizumab, autoimmune, inflammation.
Introduction
Interleukin-6 (IL-6), initially designated as a B cell differentiation factor [1], is a representative cytokine featuring redundancy and pleiotropic activity [2-4]. In the early phase of infectious inflammation, IL-6 is produced by monocytes and macrophages immediately after the stimulation of Toll-like receptors (TLRs) with distinct pathogen-associated molecular patterns (PAMPs) [5]. In noninfectious inflammations, such as burn or traumatic injury, damage-associated molecular patterns (DAMPs) from damaged or dying cells stimulate TLRs to produce IL-6 [6]. This acute IL-6 expression plays a central role in host defense by stimulating various cell populations. When acting on hapatocytes, IL-6 strongly induces a broad spectrum of acute-phase proteins such as C-reactive protein (CRP), serum amyloid A (SAA), fibrinogen, hepcidin, haptoglobin, and antichymotrypsin, whereas it reduces albumin, cytochrome P 450, fibronectin, and transferrin [7, 8] (Figure 1).
Figure 1
IL-6 has a pleiotropic effect but its dysregulated persistent production causes the onset and development of various autoimmune and chronic inflammatory diseases. IL-6 is originally found as a B cell stimulatory factor-2, which induces activated B cells into antibody production. IL-6, combined with TGF-β, preferentially induces the differentiation of naïve CD4 positive T cells into Th17 cells whereas IL-6 inhibits TGF-β induced regulatory T cell (Treg) development. As a consequence, Th17/Treg imbalance may cause the onset and progression of autoimmune and chronic inflammatory diseases. IL-6 induces production of acute-phase proteins such as CRP, fibrinogen, serum amyloid A, and hepcidin, whereas it reduces synthesis of albumin in hepatocytes. High persistent levels of serum amyloid A and hepcidin lead to amyloid A amyloidosis and anemia of inflammation, respectively. In bone marrow, IL-6 induces maturation of megakaryocytes into platelets and activation of hematopoietic stem cells. In addition, IL-6 promotes the differentiation of osteoclasts and angiogenesis, the proliferation of keratinocytes and mesangial cells, and the growth of myeloma and plasmacytoma cells. Treg: regulatory T cells; CRP: C-reactive protein; VEGF: vascular endothelial growth factor; RANKL: receptor activator of NF-kappaB ligand.
Int J Biol Sci Image (Click on the image to enlarge.)
CRP is a good biomarker of inflammation and is used as such in clinical laboratory tests. Its expression mainly depends on IL-6 [9]. If the free concentration of the anti-interleukin 6 receptor antibody, tocilizumab is maintained in serum at more than 1 μg/ml, CRP remains negative [10], so that the serum CRP level is a hallmark for checking whether IL-6 activity is completely blocked in vivo. Continuously high levels of hepcidin induced by IL-6 block iron transporter ferroportin 1 in macrophages, hepatocytes, and gut epithelial cells and lead to hypoferremia and anemia of chronic inflammation [11], whereas long-term high levels of SAA result in amyloid A amyloidosis [12]. In lymphocytes, IL-6 induces B cell differentiation into immunoglobulin-producing cells [1]. When CD4-positive naïve T cells are primed, a specific cytokine prompts their differentiation into an effector T cell subset. IL-6 together with TGF-β preferentially promotes differentiation of IL-17-producing T helper cells (Th17) that play a crucial role in the induction of autoimmune tissue injury, whereas IL-6 inhibits TGF-β-induced regulatory T cell (Treg) differentiation [13, 14]. The resultant Th17/Treg imbalance leads to breakage of immunological tolerance and is of pathological importance for the development of various autoimmune and chronic inflammatory diseases [15]. IL-6 also induces CD8-positive T cells to generate cytotoxic T cells [16]. The function of IL-6 in hematopoiesis is to induce maturation of megakaryocytes into platelets as well as activation of hematopoietic stem cells [17]. IL-6 production in bone marrow stromal cells generates the receptor activator of NF-kappaB ligand (RANKL), which is an essential factor for the differentiation and activation of osteoclasts and bone resorption, thus leading to osteoporosis [18]. Enhanced angiogenesis and increased vascular permeability are pathological features of inflammation, and these characteristics are due to the excess production of vascular endothelial growth factor (VEGF), which is induced by IL-6 in inflamed lesions such as seen in synovium tissue of rheumatoid arthritis [19]. The promotional activities of IL-6, such as the proliferation of keratinocytes or collagen production in dermal fibroblasts, may contribute to autoimmune skin diseases including psoriasis and systemic sclerosis [20, 21]. Furthermore, IL-6 stimulates the growth of cells such as myeloma/plasmacytoma cells and mesangial cells [22-24].
IL-6 triggers signal transduction after binding to the IL-6 receptor (IL-6R) [25, 26]. There are two forms of IL-6R, a transmembrane 80-kDa form with a short cytoplasmic domain and a soluble form (sIL-6R). After binding of IL-6 to transmembrane IL-6R, the resultant IL-6/IL-6R complex associates with gp130 [27-29], and the activated IL-6 receptor complex is formed as a hexameric structure consisting of two molecules each of IL-6, IL-6R and gp130 (so-called a classical signaling) [30, 31]. The expression of transmembrane IL-6R is limited to a few cell types but the IL-6/sIL-6R complex can also transduce the IL-6 signal to various cells which do not express transmembrane IL-6R but express gp130 (known as a trans-signaling mechanism) [32], so that IL-6 affects a wide variety of cells.
Pathological Role of IL-6 in Development of Diseases
When IL-6 is synthesized transiently, it promptly participates in the host defense against environmental stress such as infection and injury and at the same time provides an SOS (warning) signal by triggering a broad spectrum of biological events. Once the source of stress is removed from the host, IL-6-mediated activation of the signal transduction cascade is terminated by negatively regulatory systems in conjunction with the normalization of serum IL-6 and CRP levels. However, dysregulated persistent IL-6 production has been implicated in the development of various autoimmune, chronic inflammatory diseases and even cancers [2-4, 33]. The reason(s) why such dysregulated continuous IL-6 production is induced remains to be clarified and elucidation of the mechanism(s) underlying persistent IL-6 synthesis in diseases is of particular importance to make their pathogenesis clear. It was found that in human immunodeficiency virus (HIV)-positive cases of multicentric Castleman's diseases all patients were infected with the Kaposi sarcoma-associated herpes virus (KSHV) and that sustained synthesis of both virus-derived IL-6, which directly binds to and stimulates human gp130, and of host-derived human IL-6 contribute to the development of the disease [34].
Moreover, numerous animal models of diseases have also disclosed the pathologic role of IL-6 in disease development and that IL-6 blockade by means of gene-knockout or administration of anti-IL-6 or anti-IL-6R antibody can suppress such disease development either preventively or therapeutically. For example, IL-6 blockade strategy demonstrably limited susceptibility to Castleman's disease-like symptoms in IL-6 transgenic mice [35], as well as in various mouse models of rheumatoid arthritis [36-48], systemic lupus erythematosus [49-51], scleroderma [52, 53], C-peptide-induced myositis [54], experimental autoimmune uveoretinitis [55, 56], experimental autoimmune encephalomyelitis [57], and many other diseases.
Targeting IL-6: All the Way to Treat Autoimmune and Inflammatory Diseases
Because of the pathological role of IL-6 in various diseases, blockade of IL-6 was expected to constitute a novel treatment strategy for these diseases [3, 58, 59]. Consequently, a humanized anti-human IL-6R monoclonal antibody (chemical name: tocilizumab, generic name: Actemra outside of the EU or RoActemra inside the EU) was developed, by grafting the complementarity-determining regions of a mouse anti-human IL-6R antibody onto human IgG1. The resultant tocilizumab then blocks IL-6-mediated signal transduction by inhibiting IL-6 binding to transmembrane and soluble IL-6 receptors.
Clinical trials of tocilizumab have demonstrated its outstanding efficacy for rheumatoid arthritis [60-66], systemic juvenile idiopathic arthritis [67-71] and Castleman's disease [72, 73]. For patients with moderately to severely active rheumatoid arthritis, tocilizumab is now being used as an innovative drug in more than 90 countries worldwide. As a monotherapy or in combination with disease-modifying antirheumatic drugs, it has significantly suppressed the disease activity and radiographically detected progression of joint deformity, thus improving daily functional activity. Tocilizumab was also approved as the first line biologic for the treatment of systemic juvenile idiopathic arthritis in Japan, India, USA and EU and for Castleman's disease in Japan and India.
Furthermore, favorable results of recent pilot studies, case series or case studies have suggested that tocilizumab may have broad application for other diseases. These diseases include systemic autoimmune diseases such as systemic lupus erythematosus [74-76], systemic sclerosis [77], polymyositis [78], vasculitis syndrome including giant cell arteritis [79-84], Takayasu arteritis [79, 82, 85-87], cryoglobulinemia [88], myeloperoxidase-antineutrophil cytoplasmic antibody-associated crescentic glomerulonephritis [89] and rheumatoid vasculitis [90]. The application of tocilizumab may also extend to organ-specific autoimmune diseases including Crohn's disease [91], relapsing polychondritis [92, 93], acquired hemophilia A [94], autoimmune hemolytic anemia [95, 96], as well as to chronic inflammatory diseases such as adult-onset Still's disease [97-113], amyloid A amyloidosis [114-120], polymyalgia rheumatica [79, 84, 121], remitting seronegative symmetrical synovitis with pitting edema [122], Behcet's disease [123, 124], uveitis [125], graft-versus-host diseases [126, 127], and tumor necrosis factor receptor-associated periodic syndrome [128] (Table 1). Some studies have reported that tocilizumab is efficacious for spondyloarthritis [129-135], although others observed only minor effects [136-138]. In addition, tocilizumab is reportedly effective for pulmonary arterial hypertension [139-141], atopic dermatitis [142], and sciatica [143]. Finally, it was observed that during tocilizumab treatment of patients with rheumatoid arthritis, HbA1c levels and insulin resistance indices such as the homeostasis model assessment of insulin resistance (HOMA-IR) and the leptin-to-adiponectin ratio improved [144, 145], while serum levels of reactive oxygen metabolites decreased [146]. It can thus be expected that long-term tocilizumab treatment may offer protection against the progression of atherosclerosis leading to cardiovascular events [147]. Indeed, large-scale genetic analyses demonstrated a causal association between IL-6R-related pathways and coronary heart disease [148, 149], and a randomized, open-label, parallel-group, multicenter study to evaluate the rate of cardiovascular events of tocilizumab in comparison to a TNF inhibitor, etanercept in patients with rheumatoid arthritis (ClinicalTrials.gov, Identifier: NCT01331837) is now in progress. To establish broad clinical indications for tocilizumab for various diseases, however, further clinical studies will be needed to verify its efficacy and safety. The current clinical trials are listed in Table 2.
Table 1
Application of IL-6 blockade strategy for various autoimmune and chronic inflammatory diseases.
Approved or candidate diseases References
RA > 90 countries worldwide* 60-66
Systemic JIA Japan, India, USA and EU* 67-71
Castleman's disease Japan and India* 72,73
SLE Phase I, case reports 74-76
Systemic sclerosis Case series 77
Polymyositis Case series 78
Vasculitis syndrome Case report & series 79-90
Crohn's disease Pilot randomized trial 91
Relapsing polychondritis Case reports 92,93
Acquired hemophilia A Case report 94
Autoimmune hemolytic anemia Case reports 95,96
Adult-onset Still's disease Case reports & series 97-113
Amyloid A amyloidosis Case reports 114-120
Polymyalgia rheumatica Case reports & series 79,84,121
RS3PE Case report 122
Behcet's disease Case reports 123,124
Uveitis Case report 125
GVHD Case report & series 126,127
TRAPS Case report 128
Spondyloarthritides Case reports 129-135
Pulmonary arterial hypertension Case reports 139-141
Atopic dermatitis Case series 142
Sciatica Case series 143
Tocilizumab, a humanized anti-IL-6 receptor antibody, has been approved* as a biological drug for the treatment of RA, Castleman's disease and systemic JIA, and is expected to be applicable to various other autoimmune and chronic inflammatory diseases. RA: rheumatoid arthritis; JIA: juvenile idiopathic arthritis; SLE: systemic lupus erythematosus; RS3PE: remitting seronegative, symmetrical synovitis with pitting edema; GVHD: graft-versus-host disease; TRAPS: tumor necrosis factor-associated periodic syndrome.
Table 2
Clinical trials of tocilizumab for diseases other than RA.
Targeted diseases Identifier
ClinicalTrials.gov (USA)
Adult-onset Still's disease NCT01002781
Relapsing polychondritis NCT01041248
Type II diabetes, obesity NCT01073826
Ankylosing spondylitis NCT01209702
Graves' ophthalmopathy NCT01297609
Cardiovascular disease in rheumatoid arthritis NCT01331837
Polymyalgia rheumatica NCT01396317
Giant cell arteritis NCT01450137
Acute GVHD NCT01475162
Non-ST elevation myocardial infarction NCT01491074
Systemic sclerosis NCT01532869
Transplant rates in highly sensitized patients awaiting kidney transplantation NCT01594424
EU Clinical Trials Registry
Ankylosing spondylitis 2009-017488-40
2009-017443-34
Cardiovascular disease in rheumatoid arthritis 2010-020065-24
Graves' ophthalmopathy 2010-023841-31
Systemic sclerosis 2011-001460-22
UMIN-CTR clinical trials (Japan)
ANCA-associated vasculitis UMIN000002892
Systemic sclerosis UMIN000005550
Neuromyelitis optica UMIN000005889
UMIN000007866
Chronic glomerulonephritis UMIN000006080
Colorectal cancer UMIN000007493
Takayasu arteritis UMIN000007845
Current clinical trials of tocilizumab in the USA, EU and Japan are listed. GVHD: graft-versus-host disease; ANCA: anti-neutrophil cytoplasmic antibody.
On the basis of the pathologic role of IL-6 and the outstanding beneficial effect of tocilizumab, targeting IL-6 is a rational strategy for the treatment of various diseases and other biologics of IL-6 inhibitors are also being developed [32]. These include fully human anti-IL-6R, anti-IL-6R nanobody, anti-IL-6 antibody, and anti-IL-6/anti-IgG avimer protein consisting of the IgG-binding domain fused to the N-terminus of a 3-domain IL-6 binding region, which results in a 19-kDa heterotetrameric avimer. These novel biologics block IL-6-mediated both classical and trans-signaling pathway by inhibiting IL-6 binding to both transmembrane and soluble IL-6R. By contrast, the fusion protein soluble gp130-Fc selectively targets IL-6/sIL-6R trans-signaling pathway. It is hypothesized that IL-6 trans-signaling is a local and temporal danger signal with fewer and less important physiological functions under non-stressed conditions than classical signaling on the basis of several animal models [32, 150].
Future Directions
IL-6 participates in the host defense against environmental pathogens, whereas dysregulation of IL-6 production has been implicated in the development of various autoimmune and chronic inflammatory diseases [2-4, 33]. The pleiotropic activity of IL-6 also indicates that IL-6 blockade represents a rational treatment strategy for various diseases. A good example of the efficacy of such treatment is the dramatic improvement engendered by tocilizumab in amyloid A amyloidosis and anemia of inflammation through inhibition of their respective responsible proteins, SAA and hepcidin synthesis [151, 152]. However, the mechanisms through which tocilizumab exerts its therapeutic effects on various phenotypically different autoimmune and inflammatory diseases are not yet well understood. In recent years, it has been shown that Th17 and/or Th1 >> Treg causes the onset of various autoimmune and chronic inflammatory diseases [14, 15]. IL-6, in combination with TGF-β, promotes the differentiation of naïve T cells into Th17, but inhibits TGF-β-induced Treg differentiation, indicating that IL-6 is a very important factor for determining the Th17/Treg balance [14]. Dysregulated IL-6 production leads to predominance of Th17 over Treg but anti-IL-6R antibody can repair this imbalance. It has been demonstrated in several animal disease models that IL-6 blocking suppresses antigen-specific Th17 and/or Th1 differentiation but induces antigen-specific Treg [47, 48, 55-57]. Furthermore, it has been shown that tocilizumab in fact corrects Th17/Treg imbalance in rheumatoid arthritis patients [153]. In another study, it was found that tocilizumab induced a significant reduction in the peripheral pre-switch and post-switch memory B cells of rheumatoid arthritis patients [154] and that tocilizumab but not the TNF inhibitor significantly reduced somatic hypermutation in immunoglobulin gene rearrangements in pre-switch memory B cells [155], thus suggesting that modulation of memory B cells may be one possible target for tocilizumab. Moreover, tocilizumab treatment led to a reduction in the pathologic CD38highCD19lowIgDnegative plasma cells of SLE patients [74] and could lessen the survival of plasmablasts, which produce the anti-aquaporin 4 antibody in neuromyelitis optica [156]. These findings suggest that the clinical effect of tocilizumab is also mediated through its inhibition of pathological autoantibody production.
Because of the therapeutic efficacy of tocilizumab, IL-6 plays a major role in the onset or development of various phenotypically different diseases. IL-6 is produced by a panoply of cells including monocytes, macrophages, dendritic cells, T and B cells, neutrophils, mast cells, fibroblasts, synovial cells, keratinocytes, endothelial cells, stromal cells, mesangial cells, glial cells, neurons, chondrocytes, osteoblasts, smooth muscle cells, and others in response to various stimuli [2-4, 33]. Such phenotypic difference of diseases is conceivably due to differences in cells which generate IL-6 through abnormal transcriptional activation of the IL-6 gene [157, 158] and/or inhibition of IL-6 mRNA degradation [159, 160], or dose-dependent effects of IL-6 produced by cells recruited into the organs. Some virus products from KSHV, human immunodeficiency virus (HIV), human lymphotropic virus-1 (HTLV-1) and hepatitis B virus have been reported to affect IL-6 gene activation and/or mRNA degradation [161-168]. Therefore clarification of the cell source of IL-6 production and of the mechanism(s) through which dysregulated continuous IL-6 synthesis is induced constitutes an important issue for future studies into the pathogenesis of diseases.Targeting Interleukin-6: All the Way to Treat Autoimmune and Inflammatory Diseases
https://www.ijbs.com/v08p1227.htm
Toxicology in Vitro
The ROS-mediated activation of IL-6/STAT3 signaling pathway is involved in the 27-hydroxycholesterol-induced cellular senescence in nerve cellspanelJiaoLiua1YunLiua1JuanChenaChunyanHuaMengyingTengaKailinJiaoaZhaoxiaShenaDongmeiZhuaJiaYuebZhongLiaYuanLia
a
Department of Nutrition and Food Hygiene, School of Public Health, Nanjing Medical University, Nanjing 211166, China
b
Department of Nutrition and Food Hygiene, School of Public Health, Gansu University of Chinese Medical, Lanzhou 730000, China
南京医科大学公共卫生学院营养与食品卫生系,江苏南京211166
甘肃中医药大学公共卫生学院营养与食品卫生系,甘肃兰州730000
Highlights
•
Distinguishing from Classical 17β-estradiol (E2), 27HC induced cellular senescence in nerve cells.
•
27HC induced cellular senescence in nerve cells which may be triggered by ROS production.
•
27HC-induced ROS production mediated IL-6/STAT3 signaling.
•
IL-6/STAT3 signaling was involved in 27HC-induced cellular senescence in nerve cells.
Abstract
The oxysterol 27-hydroxycholesterol (27HC) is a selective estrogen receptor modulator (SERMs), which like endogenous estrogen 17β-estradiol (E2) induces the proliferation of ER-positive breast cancer cells in vitro. Interestingly, the observation that 27HC induces adverse effects in neural system, distinguishing it from E2. It has been suggested that high levels of circulating cholesterol increase the entry of 27HC into the brain, which may induce learning and memory impairment. Based on this evidence, 27HC may be associated with neurodegenerative processes and interrupted cholesterol homeostasis in the brain. However, the biological events that participate in this process remain largely elusive. In the present study, we demonstrated that 27HC induced apparent cellular senescence in nerve cells. Senescence-associated β-galactosidase (SA-β-Gal) assay revealed that 27HC induced senescence in both BV2 cells and PC12 cells. Furthermore, we demonstrated that 27HC promoted the accumulation of cellular reactive oxygen species (ROS) in nerve cells and subsequently activation of IL-6/STAT3 signaling pathway. Notably, treatment with the ROS scavenger N-acetylcysteine (NAC) markedly blocked 27HC-induced ROS production and activation of IL-6/STAT3 signaling pathway. Either blocking the generation of ROS or inhibition of IL-6/STAT3 both attenuated 27HC-induced cellular senescence. In sum, these findings not only suggested a mechanism whereby 27HC induced cellular senescence in nerve cells, but also helped to recognize the 27HC as a novel harmful factor in neurodegenerative diseases.
The ROS-mediated activation of IL-6/STAT3 signaling pathway is involved in the 27-hydroxycholesterol-induced cellular senescence in nerve cells - ScienceDirect
https://www.sciencedirect.com/science/article/abs/pii/S0887233317301984
Disulfide bond structures of IgG molecules
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3338938/
Cysteine - the Most Important Building Block for Cellular ...
www.immunehealthscience.com/cysteine.html
Cysteine is a sulfur-containing amino acid that contributes to the sulfhydryl group in the glutathione molecule. This makes cysteine the most crucial of the three building blocks for glutathione. This means that the level of cysteine in your system is the limiting factor in how fast you can produce glutathione and how much of it you can make.N-Acetylcysteine as an antioxidant and disulphide breaking ...
https://www.ncbi.nlm.nih.gov/pubmed/29742938
As well as being involved in the antioxidant mechanism, the disulphide breaking activity of NAC also explains its mucolytic activity which is due to its effect in reducing heavily cross-linked mucus glycoproteins. Chemical features explaining the efficient disulphide breaking activity of NAC are also explained. PMID: 29742938 [Indexed for MEDLINE]
Cited by: 50
Publish Year: 2018
Author: Giancarlo Aldini, Alessandra Altomare, Giovanna Bar
Effects of N-acetyl cysteine, vitamin E and vitamin C on ...
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6690152
Jun 28, 2019 · GSH has an important role in detoxification and it can help regenerating some antioxidants such as vita-min C and E, which are also capable of detoxifying hydrogen peroxide . In addition to them, N-acetyl cysteine (NAC) is another antioxidant molecule.
Author: Esin Akbay, Beril Erdem, Ahmet Ünlü, Ahmet Barış Durukan, Mehmet Ali Onur
Publish Year: 2019
The effect of ginger extracts on the antioxidant capacity ...
https://www.sciencedirect.com/science/article/abs/pii/S1871141313000747
Total levels of antioxidant and phenolic compounds were significantly increased in the plasma of the sows and piglets by GE supplementation. Immunoglobulin G (IgG) concentrations were significantly increased in both the plasma of sows and piglets and in colostrum by 0.5% GE supplementation.
Cited by: 4
Publish Year: 2013
Author: Sung Dae Lee, Jun Ho Kim, Hyun Jung Jung, Young
Free Radic Res. 2018 Jul;52(7):751-762. doi: 10.1080/10715762.2018.1468564. Epub 2018 May 9.
N-Acetylcysteine as an antioxidant and disulphide breaking agent: the reasons why.
Aldini G1, Altomare A1, Baron G1, Vistoli G1, Carini M1, Borsani L2, Sergio F3.
Author information1
a Department of Pharmaceutical Sciences , Università degli Studi di Milano , Milan , Italy.
2
b Global Medical Information, Zambon S.p.A. , Bresso , Italy.
3
c Global Respiratory Medical Affairs, Zambon S.p.A. , Bresso , Italy.
Abstract
The main molecular mechanisms explaining the well-established antioxidant and reducing activity of N-acetylcysteine (NAC), the N-acetyl derivative of the natural amino acid l-cysteine, are summarised and critically reviewed. The antioxidant effect is due to the ability of NAC to act as a reduced glutathione (GSH) precursor; GSH is a well-known direct antioxidant and a substrate of several antioxidant enzymes. Moreover, in some conditions where a significant depletion of endogenous Cys and GSH occurs, NAC can act as a direct antioxidant for some oxidant species such as NO2 and HOX. The antioxidant activity of NAC could also be due to its effect in breaking thiolated proteins, thus releasing free thiols as well as reduced proteins, which in some cases, such as for mercaptoalbumin, have important direct antioxidant activity. As well as being involved in the antioxidant mechanism, the disulphide breaking activity of NAC also explains its mucolytic activity which is due to its effect in reducing heavily cross-linked mucus glycoproteins. Chemical features explaining the efficient disulphide breaking activity of NAC are also explained.
KEYWORDS:
Antioxidant; N-acetylcysteine; disulphide breaking agent; glutathione precursor; oxidative stressN-Acetylcysteine as an antioxidant and disulphide breaking agent: the reasons why. - PubMed - NCBI
https://www.ncbi.nlm.nih.gov/pubmed/29742938
Kardiochir Torakochirurgia Pol. 2019 Jul; 16(2): 88–92.
Effects of N-acetyl cysteine, vitamin E and vitamin C on liver glutathione levels following amiodarone treatment in rats
Esin Akbay,1 Beril Erdem,1 Ahmet Ünlü,2 Ahmet Barış Durukan,corresponding author1,2 and Mehmet Ali Onur1
1Department of Cardiology, Faculty of Science, Hacettepe University, Ankara, Turkey
2Department of Cardiovascular Surgery, Medical Park Usak Hospital, Usak, Turkey
Abstract
Introduction
Amiodarone, a pharmaceutical extensively used to suppress atrial and ventricular tachyarrhythmias, is also known to cause many side effects on many tissues. N-acetyl-cysteine (NAC), vitamin E and vitamin C are known as antioxidants for their ability to minimize oxidative stress. In the peer-reviewed literature, there is no study reporting on the protective effects of these antioxidant agents against its hepatotoxicity.
Aim
We investigated the oxidative effects of NAC, vitamins E and C on liver tissue after amiodarone treatment.
Material and methods
Rats were randomly assigned to: control; amiodarone group; amiodarone + NAC treated group; amiodarone + Vit. E group and amiodarone + Vit. C group. Liver tissues were isolated from animals and total glutathione levels were measured.
Results
In all time intervals, the level of glutathione increased. When all time intervals were compared, the amiodarone group revealed the lowest levels. The antioxidant co-administered group was studied; the glutathione levels were statistically significantly higher than the sole amiodarone group. When vitamins E, C or N-acetyl cysteine were examined, there was no statistically significant difference among them.
Conclusions
In this study we found that hepatotoxicity capacity of amiodarone may be reduced by taking up antioxidants. In addition, the effect documented here may be reproducible and may be applied to clinical settings.
Keywords: amiodarone, toxicity, N-acetyl cysteine, vitamin E, ascorbic acid, hepatotoxicity, ratsEffects of N-acetyl cysteine, vitamin E and vitamin C on liver glutathione levels following amiodarone treatment in rats
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6690152/
Published: 12 February 2019
Role of Reactive Oxygen Species in Inflammation: A Minireview
M. A. Chelombitko
Department of Biology, Moscow State University, Moscow, 119234, Russia
Abstract
Inflammation is a protective response of a multicellular organism to injury in order to localize, eliminate, and remove harmful stimuli as well as to recover (or replace) damaged tissues. There recently has been increasing evidence that reactive oxygen species (ROS) are involved in the initiation, progression, and resolution of the inflammatory response.Furthermore, ROS act as microbicidal agents and second messengers in the intracellular signaling. The latter function is performed via posttranslational modification of protein- associated redox-sensitive cysteine residues that can undergo oxidation.
At the same time, there is clear evidence that overproduction of ROS may result in cell and tissue injury and contribute to chronic inflammation underlying many neurodegenerative, cardiovascular, and metabolic diseases.
This review has focused on the role of ROS in the key inflammatory events (increased vascular permeability and leukocyte extravasation, respiratory burst and phagocytosis, and angiogenesis) and some events leading to the resolution of inflammation. In addition, the pathological function of ROS in oxidative stress is discussed.
活性氧在炎症中的作用:综述
m·a·Chelombitko
莫斯科国立大学生物系,莫斯科,119234
摘要
炎症是多细胞生物对损伤的一种保护性反应,目的是定位、消除和清除有害的刺激以及恢复(或替换)受损组织。最近有越来越多的证据表明活性氧(ROS)参与炎症反应的启动、进展和解决。
此外,ROS在细胞内信号中起杀微生物剂和第二信使的作用。后一种功能是通过翻译后修饰蛋白相关的氧化还原敏感半胱氨酸残基来实现的。
同时,有明确的证据表明,ROS的过量产生可能导致细胞和组织损伤,并导致许多神经退行性、心血管和代谢性疾病的慢性炎症。
本文综述了ROS在主要炎症事件(血管通透性增加和白细胞外渗、呼吸暴发和吞噬、血管生成)中的作用,以及一些导致炎症消退的事件。此外,还讨论了活性氧在氧化应激中的病理作用。Role of Reactive Oxygen Species in Inflammation: A Minireview | SpringerLink
https://link.springer.com/article/10.3103/S009639251804003X
Dendritic Cells Dendritic cells are the most important APCs for activating naive T cells, and they play major roles in innate responses to infections and in linking innate and adaptive immune responses.microbenotes.com/cells-of-the-immune-system/
IFN-β is expressed early upon viral infection and is critical for stimulating expression of other type I IFNs, inducing innate antiviral gene expression, and orchestrating adaptive immune responses [36,37,38,39]. A novel IFN-β reporter mouse that expresses luciferase from the IFN-β promoter was used to demonstrate the cell-type specificity of IFN-β production upon influenza virus infection [40•]. Infection of the reporter mice with wild-type IAV restricted IFN-β response to non-epithelial cells, as the virus antagonized the IFN response in infected epithelial cells. However, upon infection by a mutant virus that does not express IFN antagonist, IFN-β was expressed by infected epithelial cells and virus antigen-positive macrophages [40•]. In a mouse model of SARS-coronavirus infection, IFN-β is expressed predominantly by airway epithelial cells and plasmacytoid dendritic cells [34]. The early production of IFN-β by virus-infected epithelial cells is likely important for communicating with uninfected cells as well as immune cells.
Respiratory Epithelial Cells as Master Communicators during Viral Infections | SpringerLink
https://link.springer.com/article/10.1007/s40588-019-0111-8
MINI REVIEW ARTICLE
Front. Microbiol., 13 August 2018 | https://doi.org/10.3389/fmicb.2018.01862
Type I Interferon Induced and Antagonized by Foot-and-Mouth Disease Virus
Xiao-xia Ma1†, Li-na Ma2†, Qiu-yan Chang1, Peng Ma1, Lin-Jie Li1, Yue-ying Wang2, Zhong-ren Ma1* and Xin Cao1,2*
1Center for Biomedical Research, Northwest Minzu University, Lanzhou, China
2State Key Laboratory of Veterinary Etiological Biology, Lanzhou Veterinary Research Institute, Chinese Academy of Agricultural Sciences, Lanzhou, China
Viral infections trigger the innate immune system, serving as the first line of defense, and are characterized by the production of type I interferon (IFN). Type I IFN is expressed in a broad spectrum of cells and tissues in the host and includes various subtypes (IFN-α, IFN-β, IFN-δ, IFN-ε, IFN-κ, IFN-τ, IFN-ω, IFN-ν, and IFN-ζ). Since the discovery of type I IFN, our knowledge of the biology of type I IFN has accumulated immensely, and we now have a substantial amount of information on the molecular mechanisms of the response and induction of type I IFN, as well as the strategies utilized by viruses to evade the type I IFN response. Foot-and-mouth disease virus (FMDV) can selectively alter cellular pathways to promote viral replication and evade antiviral immune activation of type I IFN. RNA molecules generated by FMDV are sensed by the cellular receptor for pathogen-associated molecular patterns (PAMPs). FMDV preferentially activates different sensor molecules and various signal transduction pathways. Based on knowledge of the virus or RNA pathogen specificity as well as the function-structure relationship of RNA sensing, it is necessary to summarize numerous signaling adaptors that are reported to participate in the regulation of IFN gene activation.
Introduction
Foot-and-mouth disease virus (FMDV) belongs to the Aphthovirus genus in the Picornaviridae family, and a highly infection disease caused by FMDV is regarded as an important concern for animal health (Knight-Jones et al., 2016). During FMDV evolutionary process, high mutation rates of the viral genome and quasispecies dynamics are considered major genetic factors (Shih et al., 1992). Thus, a series of studies were conducted to examine the relationship between genetic changes of the viral genome and viral fitness and different host/viral pathogenicities. Except for positive/negative selection and the random drift of the genome (Domingo et al., 2003), synonymous codon usage patterns of the FMDV genome also dominate its host ranges and viral proteins with normal biological functions (Zhou et al., 2010a,b, 2011, 2013a,b,c; Ahn et al., 2011; Ma et al., 2013; Ma X.X. et al., 2016; Gao et al., 2014). Due to the high genetic diversity of FMDV, the measures for controlling this disease need to be developed comprehensively, including killing infected and in-contact animals, the limitation of animal movement and vaccination based on conventional vaccines or new typical ones (Robinson et al., 2016). To further improve measures involved in antiviral treatments and novel vaccines for controlling rapid FMDV spread, it is important to obtain a deep understanding of the interaction between the host and FMDV. The antiviral immune response is the major focus on resisting FMDV infection, including innate/adaptive immune activations (Golde et al., 2008; Toka and Golde, 2013). The innate immune system serves as the first line of defense for resisting viral infections. The rapid induction of type I interferon (IFN) and other antiviral cytokines at the site of infection are part of the defense involved in antiviral immunity. The type I IFN family of placental mammals comprises 9 recognized classes identified to date: IFN-α, IFN-β, IFN-δ, IFN-ε, IFN-κ, IFN-τ, IFN-ω, IFN-ν, and IFN-ζ (Krause and Pestka, 2005; Detournay et al., 2013).
Type I IFNs exhibit direct antiviral activities by inhibiting viral replication and mediating the cellular immune functions of both the innate and adaptive immune system, resulting in both early limitation of the virus and long-term immunity. However, viruses are capable of selecting various strategies to evade the host immune system and thus contributing to viral pathogenicity (Schulz and Mossman, 2004; Jackson et al., 2017; Sumner et al., 2017). For FMDV infection, type I IFNs also play important roles in counteracting viral infection represent a potent biotherapeutic method against FMDV (Rodríguezpulido et al., 2011; Borrego et al., 2017). This minireview summarizes the current knowledge on how type I IFN is resistant to FMDV infection and how FMDV counteracts type I IFN induction and signaling transduction to evade the type I IFN system of host.
Evasion of Antiviral Response of Type I IFN Mediated by FMDV
Upon viral infection, innate immune responses serving as the first line of defense against viruses can induce signal transduction involving IFN and finally the expression of type I IFN, which then contributes to an antiviral stage in the cells. However, many viruses have evolved strategies to evade the antiviral immune response and prevent IFN production. Picornavirus-induced host translation system shut-off has been known to result in IFNs and other cytokine suppression (Buenz and Howe, 2006). Due to these RNA viruses being armed with some viral proteases (i.e., Lpro, 2Apro, and 3Cpro), these proteases can target different signal transduction nodes mediated by IFN (Porter et al., 2006; Yang et al., 2007; Papon et al., 2009). FMDV is no exception and has evolved to use its production to evade the antiviral immune response against FMDV (Figure 2) and maximize viral replication and dissemination. In addition to promoting the generation of viral products with biological functions, FMDV Lpro and 3Cpro plays pivotal roles in disrupting the translational system of the host. For FMDV Lpro, this protease is associated with translocation to the nucleus and cleavage of the p65 subunit of NF-κB (de Los Santos et al., 2007; Zhu et al., 2010). Although FMDV Lpro can inhibit IRF3/7 to promote the interferon-stimulated response element (ISRE) (Wang et al., 2011a,c), IRF9 can assist type I and III IFNs to perform antiviral response against FMDV infection (Stark et al., 2010; Perezmartin et al., 2012), implying that the host has evolved multiple immune pathways to meet with the evasion immune response induced by FMDV. Most recently, it has been reported that FMDV Lpro can bind to the host transcription factor ADNP (activity dependent neuroprotective protein) to suppress IFN and ISG transcription and enhance FMDV replication (Medina et al., 2017). FMDV 3Cpro specializes in cleaving FMDV polyprotein into viral proteins with biological functions and ruins eIF4G, eIF4A and histone H3 to block the translation system of the host (Falk et al., 1990; Li et al., 2001). In addition to inhibition of cell protein generation by 3Cpro, it also disrupts the transcriptional levels of ISGs, blocks the translocation of STAT1-STAT2 complex to the nucleus and suppresses the ISKE promoter (Du et al., 2014). These biological functions of FMDV 3Cpro seem to ruin the IFN system against viral infection in the broad spectrum. Like the role of EMCV 3Cpro in impairing the interaction TANK and TRAF6-mediated NF-κB signaling (Huang et al., 2015), FMDV 3Cpro also cleaves TANK to block the non-canonical IKK complex signaling pathway, rather than FMDV Lpro only disrupting TANK binding activity. In general, protease encoded by FMDV can negatively regulate innate immune signaling by degradation of essential molecules in different pathway.
FIGURE 2
www.frontiersin.org
FIGURE 2. Evasion of antiviral response of type I IFN mediated by FMDV. FMDV Lpro can cleave the p65 subunit of NF-κB and inhibit IRF3/7, which promote the interferon-stimulated response element (ISRE). FMDV 3Cpro blocks the translocation of STAT1-STAT2 complex to the nucleus and suppresses the ISKE promoter. Recombinant VP1 can disrupt the formation of the canonical IKK complex to block NF-κB activity. VP3 can disrupt the assembly of the Jak1/STAT1 complex and inhibit IRF3 phosphorylation and dimerization. FMDV 2B can bind to RIG-I or LGP2 to impair related signal transduction and enhance viral replication. FMDV 3A protein can disrupt the transcriptional levels involved in RIG-I, MDA5, and MAVS and block the interaction between RIG-I/MDA5 and MAVS via its N-terminal region.
Apart from viral proteases of FMDV, other viral proteins play roles in viral immune evasion. VP1 and VP3 serve as viral structural proteins, which are the main components of the viral capsid (Mason et al., 2003). When the vp1 gene was over-expressed in the human HEK293 cell line, VP1 and sorcin can reduce NF-κB at the transcriptional levels and weaken type I IFN activity; in addition, recombinant VP1 can disrupt the formation of the canonical IKK complex to block NF-κB activity (Li et al., 2013; Ho et al., 2014). When the vp3 gene was over-expressed in the human HEK293T cell line, VP3 disrupted the assembly of the Jak1/STAT1 complex, suggesting FMDV VP3 impairs the type II IFN signaling pathway, inducing Jak1 degradation via a lysosomal pathway; Moreover, FMDV VP3 can inhibit IRF3 phosphorylation and dimerization, thereby contributing to the impairment of signal transduction involved in the type I IFN (Li et al., 2016a,b).
Turning to some non-structural proteins of FMDV, the 2B protein is capable of improving membrane permeability and carrying out cellular protein secretory pathway shut-off (Moffat et al., 2007). Recently, it has been reported that FMDV 2B can bind to RIG-I or LGP2 to impair related signal transduction and enhance viral replication, but the detailed mechanisms have not been established yet (Zhu et al., 2016, 2017). Enterovirus 71 2C proteins can bind to RelA (p65) and suppress IKKβ phosphorylation to disrupt the formation of the canonical IKK complex and impair NF-κB activation (Du et al., 2015; Li et al., 2016c). Since the 2C protein of picornaviruses are highly conserved (Gorbalenya et al., 1989), it is assumed that FMDV 2C should possess most of these activities involved in immune evasion. The FMDV 3A protein is a multifunctional non-structural protein in viral replication, virulence and host-specific genetic features (Mason et al., 2003; Ma X. et al., 2016; Bhatt et al., 2017; Bohórquez et al., 2017; Lotufo et al., 2017). For the role of FMDV 3A in the antiviral immune response, when 3A gene was over-expressed in the PK-15 cell line (swine) and the HEK293 cell line (human), 3A protein can disrupt the transcriptional levels involved in RIG-I, MDA5, and MAVS and block the interaction between RIG-I/MDA5 and MAVS via its N-terminal region (Li et al., 2016d). Although some studies involved in the structural and non-structural proteins of FMDV have noted that these viral proteins take part in immune evasion by disrupting various nodes along signaling transduction mediated by IFNs, the over-expressed individual protein with in vitro physical structure and biological function may not reflect the real situation in infected cells by FMDV.
Conclusion
Based on recent knowledge on IFNs, it is now well established that these cytokines function in the innate immune response against viral infection. Collectively, even though a series of recent reports strongly indicate that the host has evolved a complex network related to signal transduction induced by IFN against FMDV infection and FMDV relies on its viral proteins to suppress or ruin antiviral immune responses induced by IFN systems, the detailed and precise regulatory mechanisms need to be elucidated. This field is thus at a stage where there is an urgent need for the better understanding of both the basic biology and therapeutic antiviral activity of IFNs, and for deep investigations of how those proteins, without protease activities, derived from FMDV infection can influence and control the IFN signaling transduction in vivo.Frontiers | Type I Interferon Induced and Antagonized by Foot-and-Mouth Disease Virus | Microbiology
https://www.frontiersin.org/articles/10.3389/fmicb.2018.01862/full
J Immunol. Author manuscript; available in PMC 2013 Sep 1.
Type I Alveolar Epithelial Cells Mount Innate Immune Responses during Pneumococcal Pneumonia
Kazuko Yamamoto,*† Joseph D. Ferrari,* Yuxia Cao,* Maria I. Ramirez,* Matthew R. Jones,* Lee J. Quinton,* and Joseph P. Mizgerd*
Author information Copyright and License information Disclaimer
*Pulmonary Center, Boston University School of Medicine, Boston, MA 02118, USA
†Department of Molecular Microbiology and Immunology, Nagasaki University Graduate School of Biomedical Sciences, Nagasaki, Japan
Address correspondence and reprint requests to Dr. Joseph P. Mizgerd, Pulmonary Center, Boston University School of Medicine, 72 E. Concord Street, Boston, MA 02118, USA. phone: 617-638-5201, fax: 617-638-5227, ude.ub@dregzimj
Abstract
Pneumonia results from bacteria in the alveoli. The alveolar epithelium consists of type II cells, which secrete surfactant and associated proteins, and type I cells, which constitute 95% of the surface area and met anatomic and structural needs.Other than constitutively expressed surfactant proteins, it is unknown whether alveolar epithelial cells have distinct roles in innate immunity. Since innate immunity gene induction depends on NF-κB RelA (also known as p65) during pneumonia, we generated a murine model of RelA mutated throughout the alveolar epithelium.
In response to LPS, only 2 of 84 cytokine transcripts (CCL20 and CXCL5) were blunted in lungs of mutants, suggesting that a very limited subset of immune mediators is selectively elaborated by the alveolar epithelium. Lung CCL20 induction required epithelial RelA regardless of stimulus, whereas lung CXCL5 expression depended on RelA after instillation of LPS but not pneumococcus. RelA knockdown in vitro suggested that CXCL5 induction required RelA in type II cells but not type I cells. Sorted cell populations from mouse lungs revealed that CXCL5 was induced during pneumonia in type I cells, which did not require RelA. TLR2 and STING were also induced in type I cells, with RelA essential for TLR2 but not STING. To our knowledge, these data are the first direct demonstration that type I cells, which constitute the majority of the alveolar surface, mount innate immune responses during bacterial infection. These are also the first evidence for entirely RelA-independent pathways of innate immunity gene induction in any cell during pneumonia.
Type I Alveolar Epithelial Cells Mount Innate Immune Responses during Pneumococcal Pneumonia
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3424336/
Differential Response of Primary Alveolar Type I and Type II Cells to LPS Stimulation
Mandi H. Wong, Meshell D. Johnson
Published: January 31, 2013https://doi.org/10.1371/journal.pone.0055545
Department of Veterans Affairs (CDA2) and the Northern California Institute for Research and Education
Abstract
The alveolar epithelium serves as a barrier between organism and environment and functions as the first line of protection against potential respiratory pathogens.Alveolar type II (TII) cells have traditionally been considered the immune cells of the alveolar epithelium, as they possess immunomodulatory functions; however, the precise role of alveolar type I (TI) cells, which comprise ∼95% of the alveolar epithelial surface area, in lung immunity is not clear.
We sought to determine if there was a difference in the response of TI and TII cells to lung injury and if TI cells could actively participate in the alveolar immune response. TI cells isolated via fluorescence activated cell sorting (FACS) from LPS-injured rats demonstrated greater fold-induction of multiple inflammatory mediators than TII cells isolated in the same manner from the same animals.
Levels of the cytokines TNF-α, IL-6 and IL-1β from cultured primary rat TI cells after LPS stimulation were significantly increased compared to similarly studied primary rat TII cells. We found that contrary to published reports, cultured TII cells produce relatively small amounts of TNF-α, IL-6 and IL-1β after LPS treatment; the higher levels of cytokine expression from cultured TII cells reported in the literature were likely from macrophage contamination due to traditional non-FACS TII cell isolation methods. Co-culture of TII cells with macrophages prior to LPS stimulation increased TNF-α and IL-6 production to levels reported by other investigators for TII cells, however, co-culture of TI cells and macrophages prior to LPS treatment resulted in marked increases in TNF-α and IL-6 production.
Finally, exogenous surfactant blunted the IL-6 response to LPS in cultured TI cells. Taken together, these findings advocate a role for TI cells in the innate immune response and suggest that both TI and TII cells are active players in host defense mechanisms in the lung.
Exogenous surfactant decreases LPS-stimulated TI cell cytokine production
It has been long reported that surfactant can decrease the degree of lung inflammation in response to endotoxin stimulation [33]–[35]. These studies, coupled with our results revealing a decrease in IL-6 production in co-cultures of TII cells and macrophages treated with LPS, prompted us to investigate the role of surfactant in cytokine expression in both TI and TII cells. The addition of exogenous rat surfactant (5 or 10 µg/ml) to cultured TI cells did not alter cytokine production at baseline, but significantly blunted the cytokine response of TI cells to LPS for IL-6 (LPS: 1201.9±40.6 pg/ml; LPS +5 µg/ml surfactant: 823.6±102.4 pg/ml; LPS +10 µg/ml surfactant: 782.8±71.1 pg/ml; *p<0.05). Levels for TNF-α in TI cells treated with surfactant and LPS (LPS +5 µg/ml surfactant: 886.8±99.1 pg/ml; LPS +10 µg/ml surfactant: 892.7±142.6 pg/ml) were not significantly different than TI cells stimulated with LPS alone (1256.3±259.2 pg/ml)(Figure 7). The addition of exogenous rat surfactant (5 or 10 µg/ml) to TII cells did not alter cytokine production in response to LPS (data not shown).
Figure 7. Effect of surfactant on LPS-stimulated cytokine response in TI cells.
Surfactant significantly decreased LPS-induced IL-6 production in TI cells by 43% with 5 µg/ml surfactant (n = 4) and 35% with 10 µg/ml surfactant (n = 3) when compared to IL-6 production in TI cells stimulated with LPS alone (LPS 10 µg/ml, n = 4, *p<0.05). Surfactant also decreased the level of TNF-α production in TI cells stimulated with LPS by ∼11% at both concentrations, but the reduction was not significant. Surfactant alone did not alter TNF-α or IL-6 expression in the absence of LPS (n = 3).
Studies of pulmonary surfactant in the immune response have suggested that surfactant dampens inflammation, particularly in the face of endotoxin-stimulated lung injury [34], [46], [47], but the mechanism by which this occurs has remained unclear. Surfactant has been shown to decrease cytokine production in whole lungs [48], [49], and this general finding has been attributed to the effects of surfactant on alveolar macrophages and dendritic cells. There have been reports that surfactant may bind members of the TLR-4 pathway or may inhibit the translocation of TLR-4 to lipid raft domains, thus blunting TLR-4-driven cytokine production [33], [50]. Our co-cultures of TII cells and macrophages displayed reduced levels of IL-6 when compared to IL-6 production in LPS-treated macrophages alone, initially suggesting that surfactant may be blunting the IL-6 response. Our findings demonstrated that exogenous surfactant decreased TNF-α and IL-6 production in LPS-treated TI cells, but did not significantly affect cultured TII cells or macrophages. Given that TII cells produce surfactant, additional surfactant may not have conferred any additional biological effects. And while the doses utilized here have been used in other studies, our doses in macrophages may have not been high enough to elicit an inhibitory response [33], [35], [50]. Nevertheless, our studies suggest that there may be a component of cellular interplay between TII cells and macrophages in co-culture that is leading to a reduction in IL-6 production after LPS stimulation. Furthermore, the fact that surfactant decreased cytokine production in LPS-treated TI cells suggests that any portion of the dampened inflammatory cytokine response in the distal lung achieved through the addition of exogenous surfactant may have been due to the actions of surfactant on TI cells alone. Future work in determining how surfactant can affect the immunity of TI cells may lead to a better understanding of how surfactant modulates the immune response of the lung.
LPS-induced cytokine response in TI cells may be due to stimulation of the NF-κB/IκB pathway
The NF-κB/IκB pathway has been implicated in the inflammatory cascade caused by a variety of stimuli, including LPS, in TII cells [29], [36]–[38]. LPS, acting through the toll-like receptor 4 (TLR4), can activate the phosphorylation of IKKα/β, which allows phosphorylation of IκB within the NF-κB complex, leading to ubiquitination of the IκB complex and release of NF-κB, allowing its translocation to the nucleus where it enhances transcription of various inflammatory mediators. Whether this mechanism of inflammation was present in TI cells was unclear. Using immunohistochemistry, phospho-IKKα/β staining was present in cultured TI cells as early as 10 minutes after LPS stimulation, with a progressive reduction in staining intensity at 30 minutes, 1 hour, 4 hours, 8 hours and 18 hours (Figure 8A). Similar staining was seen in TII cells (data not shown). NF-κB expression, as measured by qPCR in cultured cells, is elevated in both TI and TII cells after LPS stimulation, suggesting that LPS may act via the well-described NF-κB/IκB pathway in both cell types to stimulate cytokine secretion (Figure 8B).The NF-κB pathway is important in stimulating pro-inflammatory cytokines and activating genes that regulate the inflammatory response in multiple different organs, tissue and cells [51]. Up-regulation of NF-κB has been shown to enhance cytokine expression in TII cells but whether a similar mechanism occurs in TI cells was not known [29], [36]–[38]. TI cells exposed to LPS for 10 minutes demonstrated phosphorylated IKKα/β staining that progressively decreased in intensity after 30 minutes, which correlates with the known rapid activation of the NF-κB pathway upon LPS stimulation [38]. In addition, NF-κB transcription was elevated in cultured TI and TII cells stimulated with LPS after 8 hours, further suggesting that this may be one method by which LPS induces cytokine expression in alveolar epithelial cells.
DISCUSION
...
Our data has shown that TI and TII cells not only respond differently to LPS stimulation, but there exists the possibility that the two alveolar cell types work in concert within their microenvironment to provide a balanced inflammatory response to infection. One could postulate that TI cells may serve to heighten the pro-inflammatory response with enhanced cytokine production, particularly in the presence of alveolar macrophages, while TII cells, through the production of surfactant and relatively modest cytokine expression, provide more of an anti-inflammatory response by trying to contain inflammation. Future investigation into possible mechanisms contributing to the differential inflammatory response of TI and TII cells to LPS, along with more characterization of the alveolar microenvironment during injury, are essential to better understanding the role of the alveolar epithelium and its component cells in the innate immune response of the distal lung. Studies to determine if the responses reported here are stimulus-specific or if the alveolar epithelial cells respond similarly to other respiratory insults such as viruses, gram-positive bacteria or tobacco smoke are also warranted. Given that diseases such as pneumonia and acute respiratory distress syndrome all heavily impact diverse patient populations, elucidating the role of TI and TII cells in the innate immune response could help develop new therapies and treatment options to improve outcomes for all affected patients.
https://doi.org/10.1371/journal.pone.0055545.g007Differential Response of Primary Alveolar Type I and Type II Cells to LPS Stimulation
https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0055545Am J Physiol Lung Cell Mol Physiol. 2010 Jun; 298(6): L715–L731.
Epithelial repair mechanisms in the lung
Lynn M. Crosby1 and Christopher M. Waterscorresponding author1,2,31Department of Cell Biology, Duke Medicine, Durham NC 27705
2Division of Pulmonary, Allergy and Critical Care Medicine, Duke Medicine, Durham NC 27705
3Division of Pulmonary and Critical Care, Department of Medicine, University of California-San Francisco, San Francisco, CA 94143
4Department of Cell and Developmental Biology, University of Pennsylvania, Philadelphia, PA 19104
5Department of Internal Medicine, University of Texas-Southwestern Medical Center, Dallas, TX 75390
6Departments of Anesthesiology and Biomedical Engineering, Yale University, New Haven, CT 06520
7Department of Cell Biology and Physiology, The University of North Carolina School of Medicine, Chapel Hill, NC 27599
8Department of Anatomy, University of California-San Francisco, San Francisco, CA 94143
9Perleman School of Medicine, University of Pennsylvania, Philadelphia, PA 19104
10Regenerative Medicine Institute, Cedars-Sinai Medical Center, Los Angeles, CA 90048
11Department of Internal Medicine, University of Michigan, Ann Arbor, MI 48109
12Section of Neonatology, Perinatal and Pulmonary Biology, Department of Pediatrics, Cincinnati Children’s Hospital Center, University of Cincinnati College of Medicine, Cincinnati, OH 45229
13Departments of Medicine and Cell and Developmental Biology, University of Pennsylvania, Philadelphia, PA 19104
*Address correspondence to: Brigid Hogan PhD, Department of Cell Biology, Room 388C, Nanaline Duke Building, 7513 Research Drive, Duke Medicine, Durham NC 27710, Phone: 919-684-8085, FAX: 919-684-8592, ude.ekud@nagoh.digirb. Edward E. Morrisey, Ph.D., University of Pennsylvania, Translational Research Center, Room 11-124, 3400 Civic Center Boulevard, Building 421, Philadelphia, PA 19104-5129, Phone: 215-573-3010, FAX: 215-573-2094, ude.nnepu.dem.liam@esirrome
Abstract
The recovery of an intact epithelium following lung injury is critical for restoration of lung homeostasis. The initial processes following injury include an acute inflammatory response, recruitment of immune cells, and epithelial cell spreading and migration upon an autologously secreted provisional matrix. Injury causes the release of factors that contribute to repair mechanisms including members of the epidermal growth factor and fibroblast growth factor families (TGF-α, KGF, HGF), chemokines (MCP-1), interleukins (IL-1β, IL-2, IL-4, IL-13), and prostaglandins (PGE2), for example. These factors coordinate processes involving integrins, matrix materials (fibronectin, collagen, laminin), matrix metalloproteinases (MMP-1, MMP-7, MMP-9), focal adhesions, and cytoskeletal structures to promote cell spreading and migration. Several key signaling pathways are important in regulating these processes, including sonic hedgehog, Rho GTPases, MAP kinase pathways, STAT3, and Wnt. Changes in mechanical forces may also affect these pathways. Both localized and distal progenitor stem cells are recruited into the injured area, and proliferation and phenotypic differentiation of these cells leads to recovery of epithelial function. Persistent injury may contribute to the pathology of diseases such as asthma, chronic obstructive pulmonary disease, and pulmonary fibrosis. For example, dysregulated repair processes involving TGF-β and epithelial-mesenchymal transition may lead to fibrosis. This review focuses on the processes of epithelial restitution, the localization and role of epithelial progenitor stem cells, the initiating factors involved in repair, and the signaling pathways involved in these processes.
Keywords: wound healing, alveolar epithelium, airway epithelium, mechanotransductionRepair and regeneration of the respiratory system: complexity, plasticity, and mechanisms of lung stem cell function
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4212493/
Nature. Author manuscript; available in PMC 2018 Aug 28.
Regeneration of the lung alveolus by an evolutionarily conserved epithelial progenitor
William J. Zacharias,1,2,# David B. Frank,2,3,4,# Jarod A. Zepp,1,2 Michael P. Morley,1,2,4 Farrah Alkhaleel,1,2 Jun Kong,1,2 Su Zhou,1,4 Edward Cantu,7 and Edward E. Morrisey1,2,4,5,6,*
Abstract
Functional tissue regeneration is required for restoration of normal organ homeostasis after severe injury. While some organs, such as the intestine, harbor active stem cells throughout homeostasis and regeneration1, more quiescent organs like the lung often contain facultative progenitor cells which are recruited after injury to participate in regeneration2,3. Here we show that a Wnt-responsive alveolar epithelial progenitor (AEP) lineage within the alveolar type 2 (AT2) cell population acts as a major facultative progenitor cell in the distal lung. AEPs are a stable lineage during alveolar homeostasis but expand rapidly to regenerate a large proportion of the alveolar epithelium after acute lung injury. AEPs exhibit a distinct transcriptome, epigenome, and functional phenotype with specific responsiveness to Wnt and Fgf signaling. In distinction to other proposed lung progenitor cells, human AEPs (hAEPs) can be directly isolated via expression of the conserved cell surface marker TM4SF1, and hAEPs act as functional human alveolar epithelial progenitor cells in 3D organoids. Together, our results identify the AEP lineage as an evolutionarily conserved alveolar progenitor and a new target for human lung regeneration strategies.Regeneration of the lung alveolus by an evolutionarily conserved epithelial progenitor
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6020060/
Curr Med Chem. 2012;19(35):6003-8.
Alveolar epithelial stem and progenitor cells: emerging evidence for their role in lung regeneration.
Hoffman AM1, Ingenito EP.
Author information
Abstract
Lung injuries that impact the alveolus, such as emphysema, pulmonary fibrosis, and acute lung injury, are costly and prevalent problems. Moreover, the extent of alveolar injury and impairment of gas exchange is strongly associated with prognosis and survival. Thus, mechanisms of repair and regeneration of the lung alveolar compartment have received mounting attention as newer approaches to the study of stem and progenitor cells in this region unfold. The role of type II alveolar epithelial as the sole source of type I (AECI) and II (AECII) alveolar epithelial cells following lung injury has been recently challenged; recently, investigators have described stemprogenitor cells that function like precursors to AECII either in vitro or in vivo, both in mice and humans. Techniques to explore selfrenewal and multipotency have been rigorously applied to these putative stem-progenitor cell populations and the data thus far is compelling. This review provides background to the study of alveolar regeneration with the aim to provide context to the recent discoveries of putative stem-progenitor cells that may contribute to this process.
PMID: 23016551 DOI: 10.2174/092986712804485872Harnessing the potential of lung stem cells for regenerative medicine. - PubMed - NCBI
https://www.ncbi.nlm.nih.gov/pubmed/25450456
Nature. 2018 Mar 8;555(7695):251-255. doi: 10.1038/nature25786. Epub 2018 Feb 28.
Regeneration of the lung alveolus by an evolutionarily conserved epithelial progenitor.
Zacharias WJ1,2, Frank DB2,3,4, Zepp JA1,2, Morley MP1,2,4, Alkhaleel FA1,2, Kong J1,2, Zhou S1,4, Cantu E5, Morrisey EE1,2,4,6,7.
Author information1
Department of Medicine, University of Pennsylvania, Philadelphia, Pennsylvania 19104, USA.
2
Penn Center for Pulmonary Biology, University of Pennsylvania, Philadelphia, Pennsylvania 19104, USA.
3
Department of Pediatrics, Children's Hospital of Philadelphia, Philadelphia, Pennsylvania 19104, USA.
4
Penn Cardiovascular Institute, University of Pennsylvania, Philadelphia, Pennsylvania 19104, USA.
5
Department of Surgery, University of Pennsylvania, Philadelphia, Pennsylvania 19104, USA.
6
Department of Cell and Developmental Biology, University of Pennsylvania, Philadelphia, Pennsylvania 19104, USA.
7
Penn Institute for Regenerative Medicine, University of Pennsylvania, Philadelphia, Pennsylvania 19104, USA.
Abstract
Functional tissue regeneration is required for the restoration of normal organ homeostasis after severe injury. Some organs, such as the intestine, harbour active stem cells throughout homeostasis and regeneration; more quiescent organs, such as the lung, often contain facultative progenitor cells that are recruited after injury to participate in regeneration.Here we show that a Wnt-responsive alveolar epithelial progenitor (AEP) lineage within the alveolar type 2 cell population acts as a major facultative progenitor cell in the distal lung. AEPs are a stable lineage during alveolar homeostasis but expand rapidly to regenerate a large proportion of the alveolar epithelium after acute lung injury. AEPs exhibit a distinct transcriptome, epigenome and functional phenotype and respond specifically to Wnt and Fgf signalling.
In contrast to other proposed lung progenitor cells, human AEPs can be directly isolated by expression of the conserved cell surface marker TM4SF1, and act as functional human alveolar epithelial progenitor cells in 3D organoids. Our results identify the AEP lineage as an evolutionarily conserved alveolar progenitor that represents a new target for human lung regeneration strategies.
Regeneration of the lung alveolus by an evolutionarily conserved epithelial progenitor. - PubMed - NCBI
https://www.ncbi.nlm.nih.gov/pubmed/29489752
Int J Biochem Cell Biol. 2014 Nov;56:82-91. doi: 10.1016/j.biocel.2014.10.012. Epub 2014 Oct 15.
Harnessing the potential of lung stem cells for regenerative medicine.
McQualter JL1, Anthony D2, Bozinovski S2, Prêle CM3, Laurent GJ3.
Author information
1
Lung Health Research Centre, Department of Pharmacology and Therapeutics, University of Melbourne, Melbourne, Vic, Australia. Electronic address: jlmcq@unimelb.edu.au.
2
Lung Health Research Centre, Department of Pharmacology and Therapeutics, University of Melbourne, Melbourne, Vic, Australia.
3
Centre for Cell Therapy and Regenerative Medicine, School of Medicine and Pharmacology, The University of Western Australia and Harry Perkins Institute of Medical Research, Perth, WA, Australia; Lung Institute of Western Australia and Centre for Asthma Allergy and Respiratory Research, School of Medicine and Pharmacology, The University of Western Australia, Perth, WA, Australia.
Abstract
In response to recurrent exposure to environmental insults such as allergens, pollution, irritants, smoke and viral/bacterial infection, the epithelium of the lung is continually damaged. Homeostasis of the lung requires a balance between immune regulation and promotion of tissue regeneration, which requires the co-ordinated proliferation and differentiation of stem and progenitor cells. In this review we reflect on the current understanding of lung epithelial stem and progenitor cells and advocate a model hierarchy in which self-renewing multipotent lung epithelial stem cells give rise to lineage restricted progenitor cells that repopulate airway and alveolar epithelial cell lineages during homeostasis and repair. We also discuss the role of mesenchymal progenitor cells in maintaining the structural integrity of the lung and propose a model in which mesenchymal cells act as the quintessential architects of lung regeneration by providing molecular signals, such as FGF-10, to regulate the fate and specificity of epithelial stem and progenitor cells. Moreover, we discuss the current status and future prospects for translating lung stem cell therapies to the clinic to replace, repair, or regenerate diseased lung tissue. This article is part of a directed issue entitled: Regenerative Medicine: the challenge of translation.
Copyright © 2014 Elsevier Ltd. All rights reserved.
KEYWORDS:
Epithelium; Lung; Mesenchyme; Regeneration; Stem cells
PMID: 25450456 DOI: 10.1016/j.biocel.2014.10.012
[Indexed for MEDLINE]Harnessing the potential of lung stem cells for regenerative medicine. - PubMed - NCBI
https://www.ncbi.nlm.nih.gov/pubmed/25450456
JCI Insight. 2020 Jan 16;5(1). pii: 131232. doi: 10.1172/jci.insight.131232.
Interleukin-13 disrupts type 2 pneumocyte stem cell act.ivity
Glisinski KM1, Schlobohm AJ1, Paramore SV1, Birukova A1, Moseley MA2, Foster MW1,2, Barkauskas CE1.
Author information
1
Division of Pulmonary, Allergy, and Critical Care Medicine, Department of Medicine, and.
2
Duke Proteomics and Metabolomics Shared Resource, Duke University Medical Center, Durham, North Carolina, USA.
Abstract
The T helper 2 (Th2) inflammatory cytokine interleukin-13 (IL-13) has been associated with both obstructive and fibrotic lung diseases; however, its specific effect on the epithelial stem cells in the gas exchange compartment of the lung (alveolar space) has not been explored. Here, we used in vivo lung models of homeostasis and repair, ex vivo organoid platforms, and potentially novel quantitative proteomic techniques to show that IL-13 disrupts the self-renewal and differentiation of both murine and human type 2 alveolar epithelial cells (AEC2s). Significantly, we find that IL-13 promotes ectopic expression of markers typically associated with bronchiolar airway cells and commonly seen in the alveolar region of lung tissue from patients with idiopathic pulmonary fibrosis. Furthermore, we identify a number of proteins that are differentially secreted by AEC2s in response to IL-13 and may provide biomarkers to identify subsets of patients with pulmonary disease driven by "Th2-high" biology.
KEYWORDS:
Adult stem cells; Proteomics; Pulmonology; Stem cells
PMID: 31941839 PMCID: PMC7030850 DOI: 10.1172/jci.insight.131232Interleukin-13 disrupts type 2 pneumocyte stem cell activity. - PubMed - NCBI
https://www.ncbi.nlm.nih.gov/pubmed/31941839
Nat Med. 2016 Nov;22(11):1285-1293. doi: 10.1038/nm.4192. Epub 2016 Oct 3.
Hyaluronan and TLR4 promote surfactant-protein-C-positive alveolar progenitor cell renewal and prevent severe pulmonary fibrosis in mice.
Liang J1,2, Zhang Y1,2, Xie T1,2, Liu N1,2, Chen H3, Geng Y1,2, Kurkciyan A1,2, Mena JM1,2, Stripp BR1,2, Jiang D1,2, Noble PW1,2.
Author information
Abstract
Successful recovery from lung injury requires the repair and regeneration of alveolar epithelial cells to restore the integrity of gas-exchanging regions within the lung and preserve organ function. Improper regeneration of the alveolar epithelium is often associated with severe pulmonary fibrosis, the latter of which involves the recruitment and activation of fibroblasts, as well as matrix accumulation.Type 2 alveolar epithelial cells (AEC2s) are stem cells in the adult lung that contribute to the lung repair process. The mechanisms that regulate AEC2 renewal are incompletely understood.
We provide evidence that expression of the innate immune receptor Toll-like receptor 4 (TLR4) and the extracellular matrix glycosaminoglycan hyaluronan (HA) on AEC2s are important for AEC2 renewal, repair of lung injury and limiting the extent of fibrosis.
Either deletion of TLR4 or HA synthase 2 in surfactant-protein-C-positive AEC2s leads to impaired renewal capacity, severe fibrosis and mortality.
Furthermore, AEC2s from patients with severe pulmonary fibrosis have reduced cell surface HA and impaired renewal capacity, suggesting that HA and TLR4 are key contributors to lung stem cell renewal and that severe pulmonary fibrosis is the result of distal epithelial stem cell failure.
Comment in
Innate immune signaling and stem cell renewal in idiopathic pulmonary fibrosis. [Nat Med. 2016]
PMID: 27694932 PMCID: PMC5503150 DOI: 10.1038/nm.4192Hyaluronan and TLR4 promote surfactant-protein-C-positive alveolar progenitor cell renewal and prevent severe pulmonary fibrosis in mice. - PubMed - NCBI
https://www.ncbi.nlm.nih.gov/pubmed/27694932
March 6, 2018
New stem cell identified for lung tissue regeneration
Mouse (left) and human (right) AEP cells grow into large lung organoids in culture and make multiple types of epithelial cells, including gas exchange type 1 cells (red) and surfactant-producing type 2 cells (green).Ed Morrisey lab, Perelman School of Medicine, University of Pennsylvania.
At a Glance
Researchers identified and characterized a stem cell that produces new air sac cells in mouse models of lung injury and in human lung tissue.
The findings uncover potentially new targets for stimulating lung regeneration.
Lung organoids
Mouse (left) and human (right) AEP cells grow into large lung organoids in culture and make multiple types of epithelial cells, including gas exchange type 1 cells (red) and surfactant-producing type 2 cells (green).Ed Morrisey lab, Perelman School of Medicine, University of Pennsylvania.
Breathing is a vital function of life. Your lungs allow your body to take in oxygen from the air and get rid of carbon dioxide, a waste gas that can be toxic.
The intake of oxygen and removal of carbon dioxide in the lung is called gas exchange. When you breathe in, air travels down your windpipe and into your lungs. After passing through your bronchial tubes, the air enters the alveoli (air sacs). Oxygen from the air passes through the very thin walls of the alveoli to the surrounding blood vessels. At the same time, carbon dioxide moves from the blood vessels into the air sacs to be exhaled.
Air sacs can be damaged from injuries, viruses, or lung disease. Damage to the air sacs can make it harder to breathe. Lung tissue is slow to regenerate. A team led by Dr. Edward E. Morrisey of the University of Pennsylvania characterized the molecular underpinnings of air sac regeneration in the lungs. The research was supported primarily by NIH’s National Heart, Lung, and Blood Institute (NHLBI). Results were published online in Nature on February 28, 2018.
The researchers compared alveoli cell activity over time in mice with lung damage from the influenza (flu) virus and healthy mice. They tracked cells that contained known markers of cells that turn into alveolar type 2 cells (AT2). AT2 cells produce the surfactant that protects alveolar type 1 (AT1) cells, which form the gas-exchange surface of the air sacs.
The team found that a month after an influenza-induced lung injury, the tracked cells rapidly expanded and produced a large increase in both AT2 and AT1 cells. The cells self-renewed and, after three months, the majority of AT2 and AT1 cells in the alveoli that had regenerated had come from the injury-induced cells, which the scientists now call alveolar epithelial progenitor (AEP) cells.
The team next characterized the gene and protein expression of the AEP cells from mouse lung. Using this information, they were able to isolate AEP cells from human lung tissue. The cells were used to grow 3D organ-like structures, called organoids, in the laboratory for further study. The researchers found molecular similarities in the cells that appear to be evolutionarily conserved between species.
“From our organoid culture system, we were able to show that AEPs are an evolutionarily conserved alveolar progenitor that represents a new target for human lung regeneration strategies,” Morrisey says.
“We are very excited at this novel finding,” says Dr. James P. Kiley, director of NHLBI’s Division of Lung Diseases. “Basic studies are fundamental stepping stones to advance our understanding of lung regeneration. Furthermore, the NHLBI support of investigators from basic to translational science helps promote collaborations that bring the field closer to regenerative strategies for both acute and chronic lung diseases."
—by Tianna Hicklin, Ph.D.New stem cell identified for lung tissue regeneration | National Institutes of Health (NIH)
https://www.nih.gov/news-events/nih-research-matters/new-stem-cell-identified-lung-tissue-regeneration
Published: 29 August 2019
Proteasome dysfunction in alveolar type 2 epithelial cells is associated with acute respiratory distress syndrome
Sneha Sitaraman, Cheng-Lun Na, Li Yang, Alyssa Filuta, James P. Bridges & Timothy E. Weaver
Scientific Reports volume 9, Article number: 12509 (2019) Cite this article
Abstract
Proteasomes are a critical component of quality control that regulate turnover of short-lived, unfolded, and misfolded proteins. Proteasome activity has been therapeutically targeted and considered as a treatment option for several chronic lung disorders including pulmonary fibrosis. Although pharmacologic inhibition of proteasome activity effectively prevents the transformation of fibroblasts to myofibroblasts, the effect on alveolar type 2 (AT2) epithelial cells is not clear. To address this knowledge gap, we generated a genetic model in which a proteasome subunit, RPT3, which promotes assembly of active 26S proteasome, was conditionally deleted in AT2 cells of mice. Partial deletion of RPT3 resulted in 26S proteasome dysfunction, leading to augmented cell stress and cell death. Acute loss of AT2 cells resulted in depletion of alveolar surfactant, disruption of the alveolar epithelial barrier and, ultimately, lethal acute respiratory distress syndrome (ARDS). This study underscores importance of proteasome function in maintenance of AT2 cell homeostasis and supports the need to further investigate the role of proteasome dysfunction in ARDS pathogenesis.Proteasome dysfunction in alveolar type 2 epithelial cells is associated with acute respiratory distress syndrome | Scientific Reports
https://www.nature.com/articles/s41598-019-49020-4
Experimental and Toxicologic Pathology
Volume 57, Issues 5–6, 10 July 2006, Pages 411-418
Experimental and Toxicologic Pathology
Heme oxygenase-1 gene expression in human alveolar epithelial cells (A549) following exposure to whole cigarette smoke on a direct in vitro exposure system
Author links open overlay panelYasuoFukanoaHiroyukiYoshimuraaTakemiYoshidab
Show more
https://doi.org/10.1016/j.etp.2005.12.001Get rights and content
Abstract
Many in vitro studies have employed cigarette smoke condensates or soluble smoke components to investigate the biological effects of cigarette smoke. However, neither of these methods evaluates the biological effects of fresh whole cigarette smoke. It is most desirable to conduct in vitro biological studies under conditions which accommodate the dynamic physicochemical character of fresh cigarette smoke.
Previously we reported the development of a whole smoke exposure system to assess the biological effects of mainstream cigarette smoke. The exposure system design was based on a combination of the sedimentation procedure and the CULTEX cultivation technique, which includes a systemized air/liquid interface methodology and exposes the cells to fresh smoke at every puff.
The aim of this study was to adopt the other biological endpoint to our whole smoke exposure system. We focused on heme oxygenase (HO)-1 mRNA gene expression, an enzyme which has recently been shown to be highly responsible for oxidative stress. In the present study, a dose–response relationship between the HO-1 mRNA expression based on the reverse transcription real-time PCR method and total exposure to cigarette smoke was observed. When a Cambridge filter pad was placed between the cigarette and exposure module, to ensure the cells were only exposed to the gas/vapor phase, the latter, as well as the whole smoke, induced HO-1 mRNA dose dependently. For the next step, acetate plain and charcoal filters with the same pressure drop were prepared to assess the potential ability of charcoal filters with regard to the vapor phase performance. The results revealed reduced HO-1 mRNA gene expression when a charcoal filter was used.
Direct whole smoke exposure is a significant approach and may reflect the conditions of exposure essentially resulting from direct contact between cells and a dynamic mixture of gaseous and particulate constituents. We were able to adopt a gene expression assay for oxidative stress to the whole smoke exposure system, following the adaptation of cytotoxicity assays. This system, which includes several advantages involving the post-exposure washing of cells, by adding the exchanging medium and assuring the exposure of the particulate phase through the sedimentation method, may have potential for further investigations into the molecular basis of smoking-related lung disease.
Previous article in issueNext article in issue
Keywords
Cigarette smokeHuman alveolar epithelial cellsWhole smoke exposureParticulate phaseVapor phaseHeme oxygenase-1Heme oxygenase-1 gene expression in human alveolar epithelial cells (A549) following exposure to whole cigarette smoke on a direct in vitro exposure system - ScienceDirect
https://www.sciencedirect.com/science/article/abs/pii/S0940299306000145
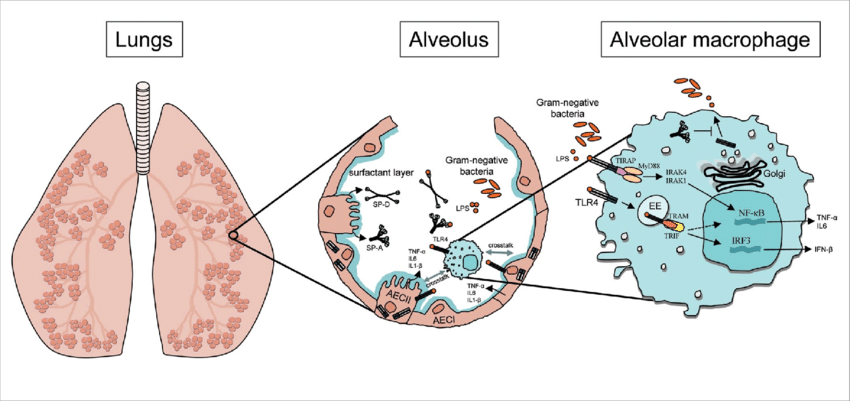
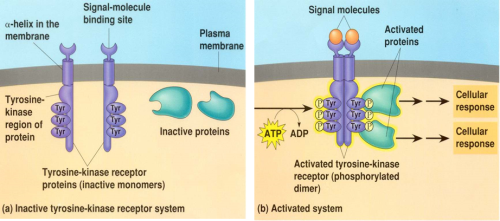
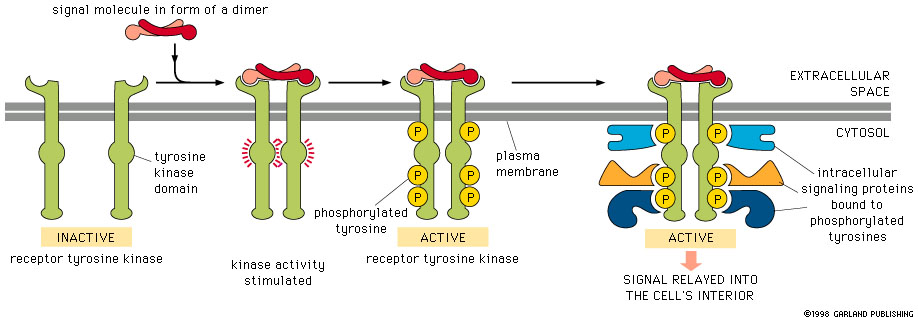


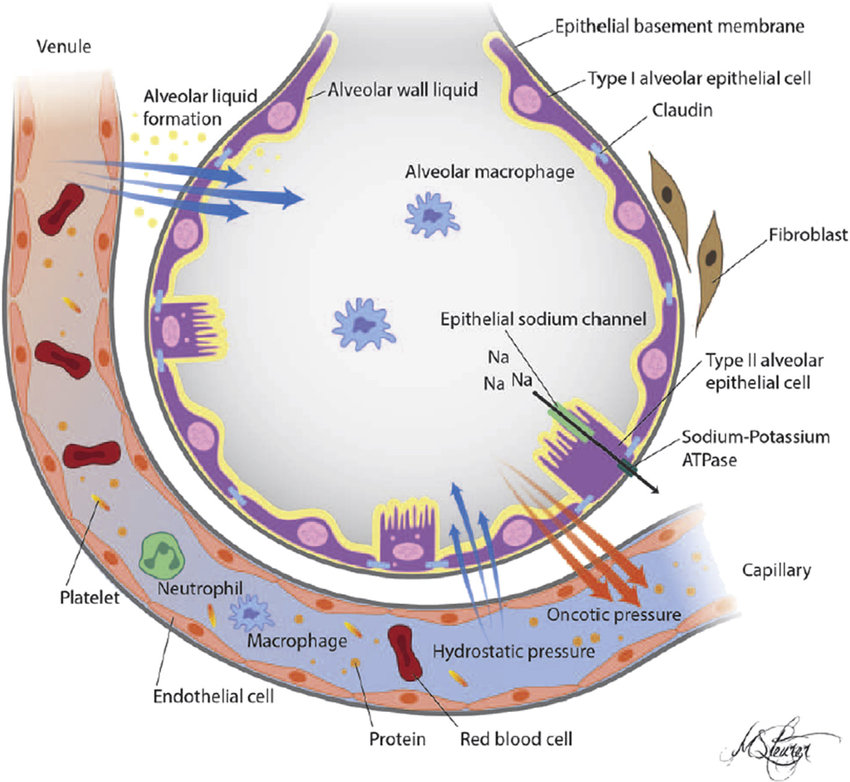

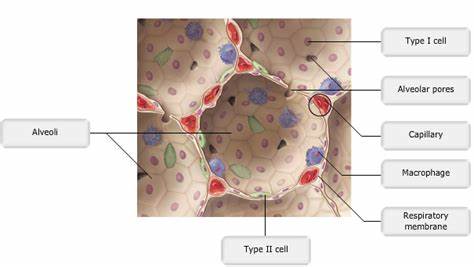
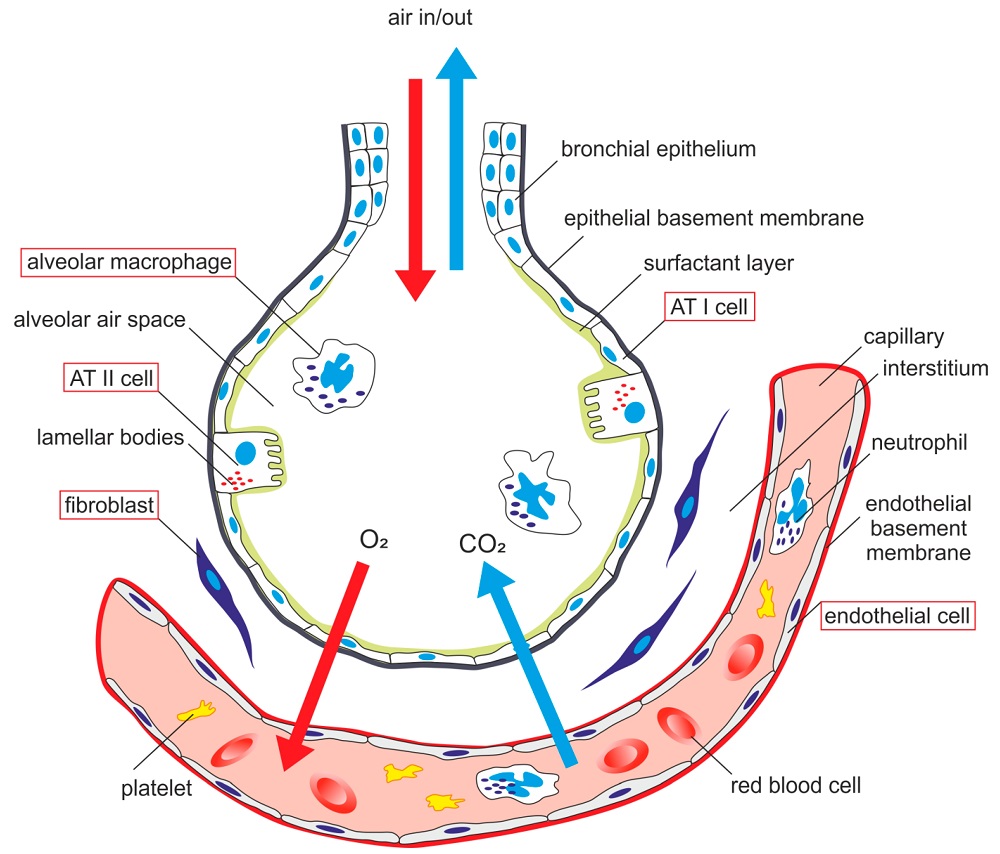
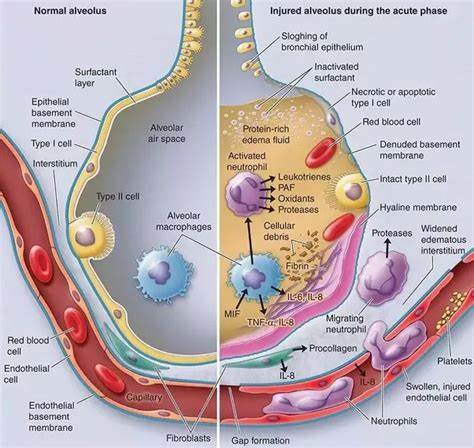
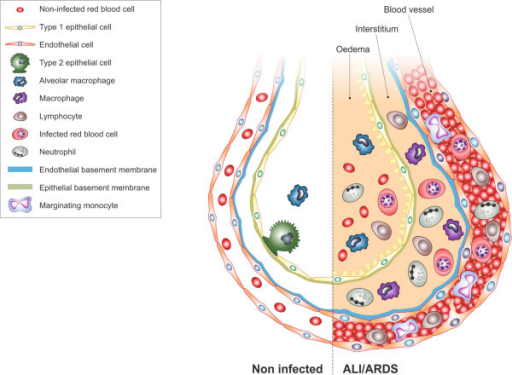

%20infection%20in%20the%20upper%20respiratory%20tract..jpg)
%20infection..jpg)
.png)
.png)
.png)
.png)
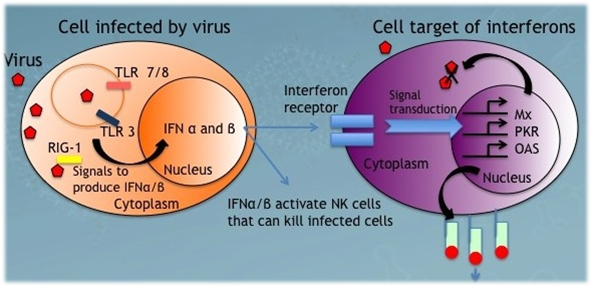
%20expression%20on%20CD44%20high%20memory%20phenotype%20CD8%20T%20cells..jpg)

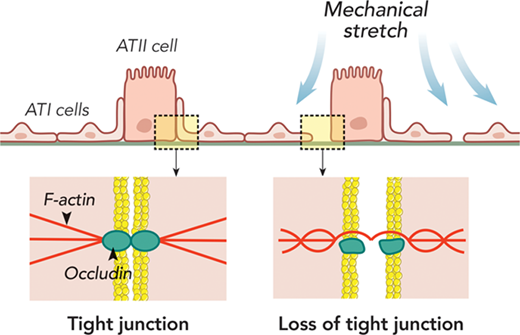

%20in%20the%20lung%20repair%20process%20in%20acute%20respiratory%20distress%20syndrome..png)

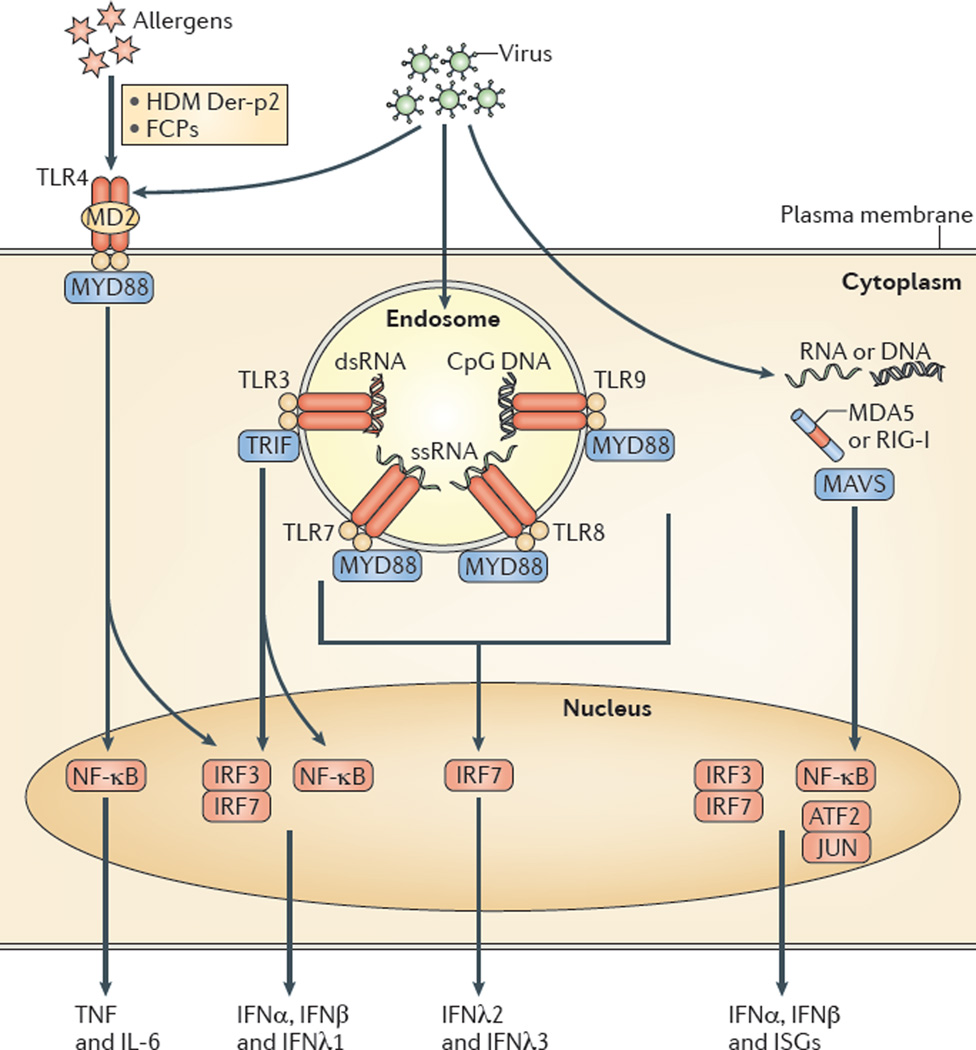
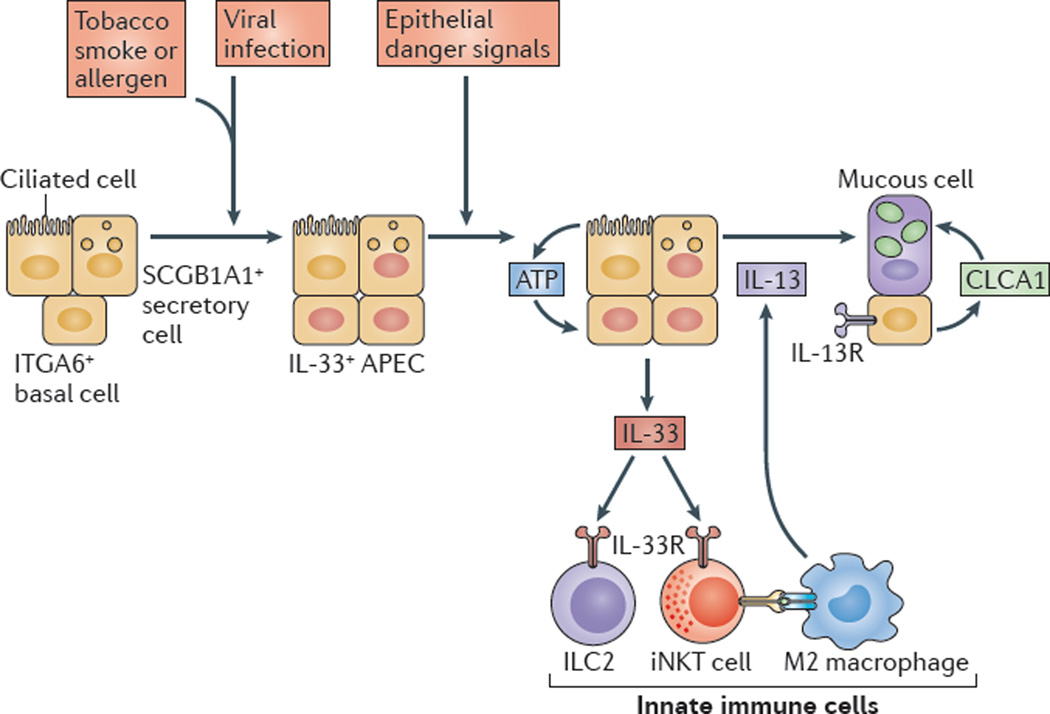
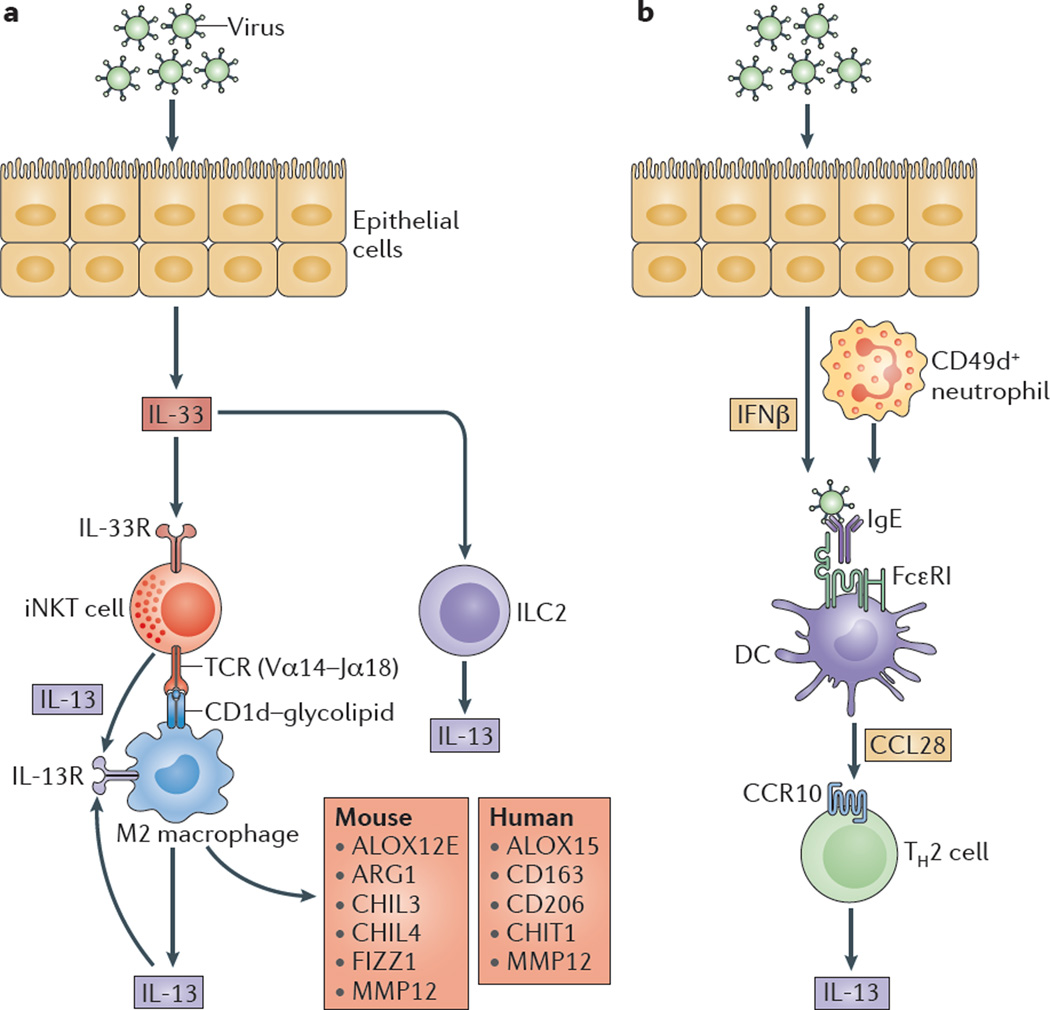
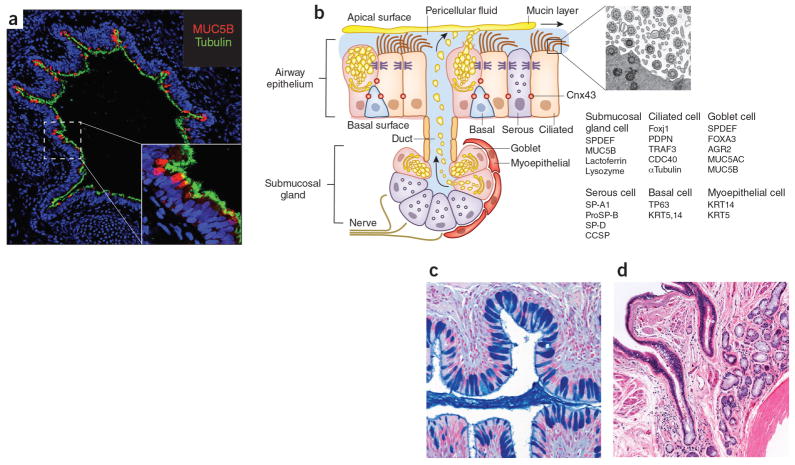
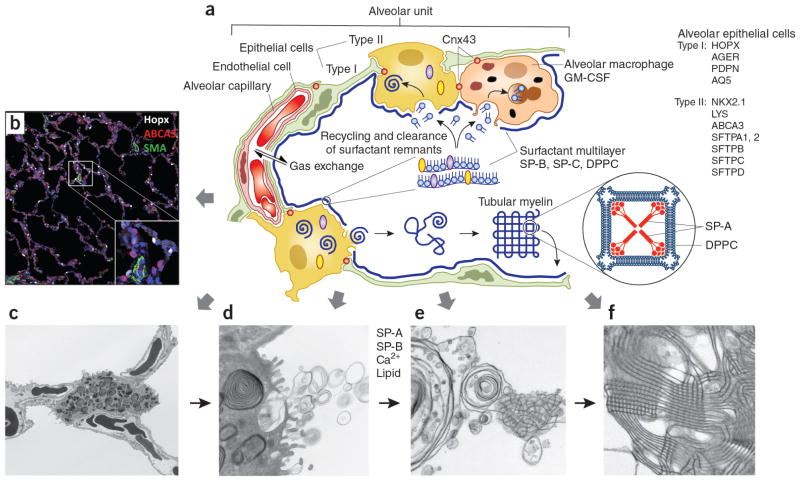
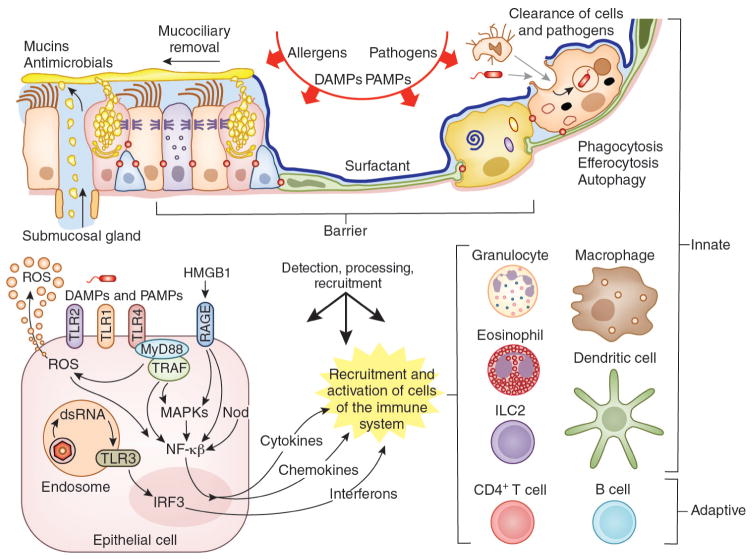
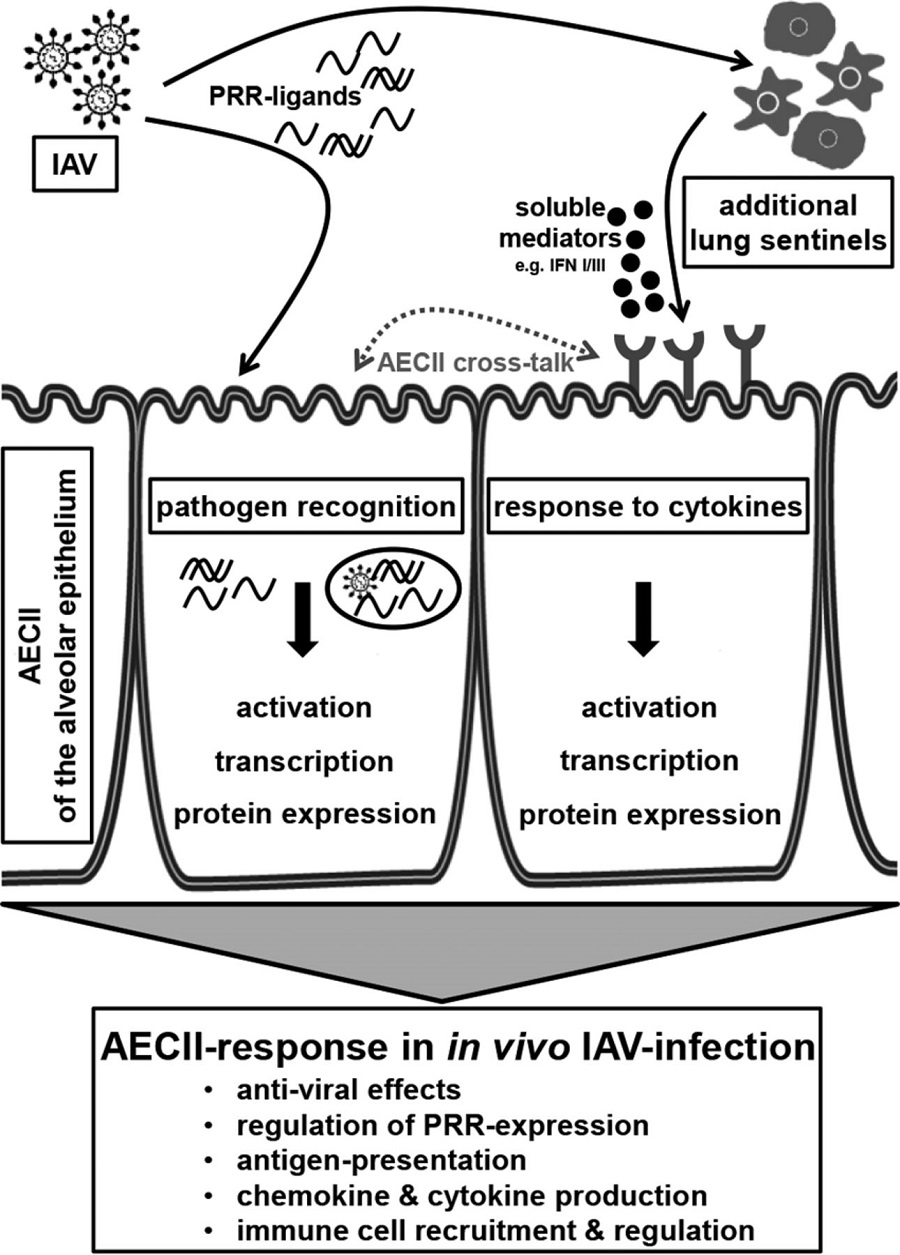
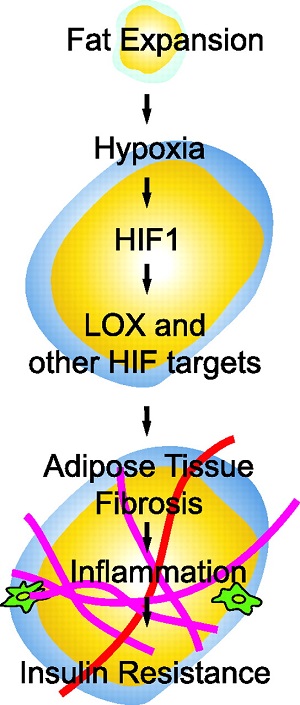
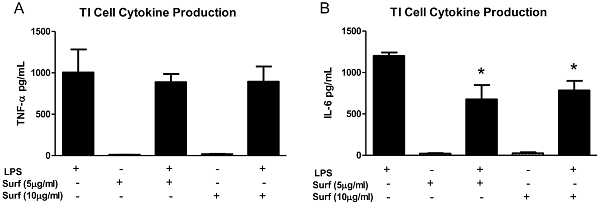

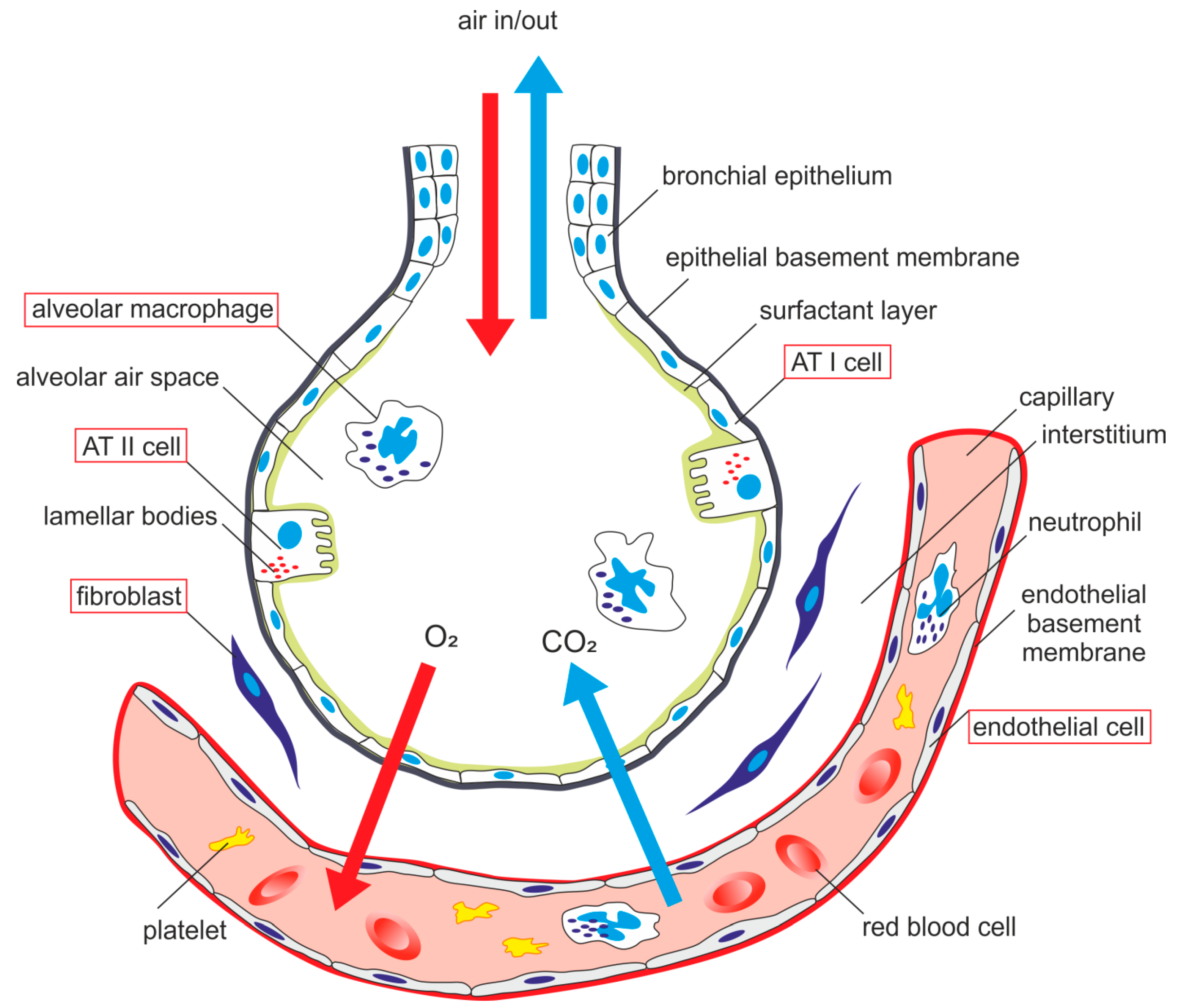{kind=link}